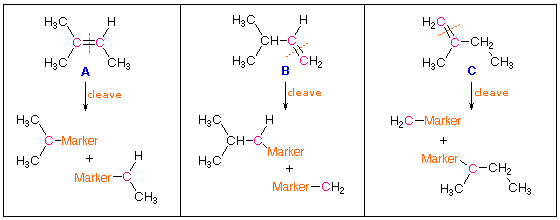
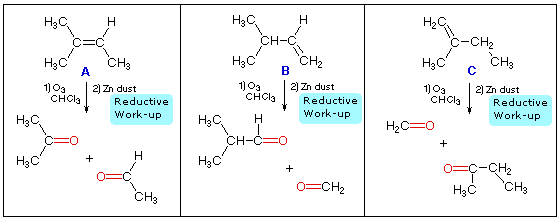
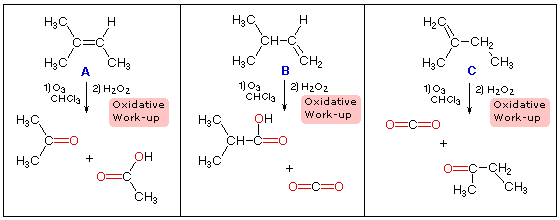
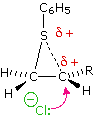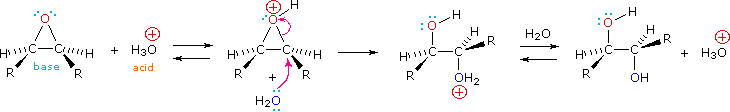quimico
2011-12-10 09:33
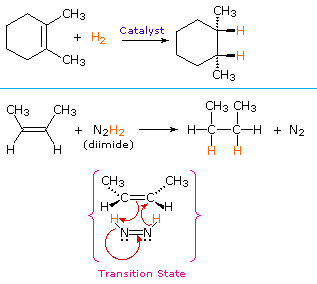
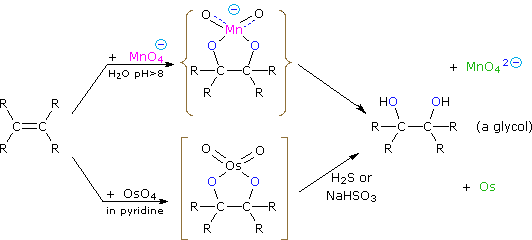
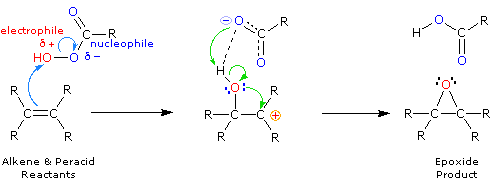
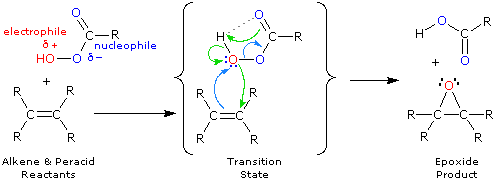
La più comune trasformazione chimica di un doppio legame carbonio-carbonio è la reazione di addizione. Un gran numero di reagenti, sia inorganici che organici, sono stati scoperti addizionarsi a questo gruppo funzionale. La maggioranza di queste reazioni sono esotermiche, a causa del fatto che il legame π C-C è relativamente debole (ca. 63 kcal/mole) rispetto ai legami σ formati da atomi o gruppi del reagente. Ricordate, le energie di legame di una molecola sono le energie richieste per rompere (omoliticamente) tutti i legami covalenti nella molecola. Di conseguenza, se le energie di legame delle molecole dei prodotti sono maggiori rispetto alle energie dei reagenti, la reazione sarà esotermica. I seguenti calcoli per l'addizione di HBr sono tipiche. Notate che per convenzione le reazioni esotermiche hanno un calore di reazione negativo.
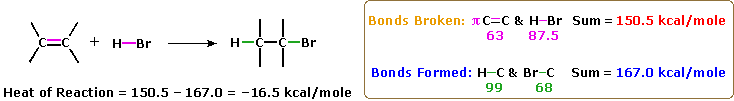 1. Addizione di forti acidi di Brønsted
Come illustrato dalle precedente equazione generale, gli acidi di Brønsted forti come HCl, HBr, HI & H2SO4, rapidamente si addizionano al gruppo funzionale C=C degli alcheni a dare prodotti in cui nuovi legami covalenti sono formati con l'idrogeno e con la base coniugata dell'acido. Usando l'equazione sopra come guida, non è difficile capire i prodotti della reazione sotto tra il cicloesene e i sopracitati acidi di Brønsted.
1. Addizione di forti acidi di Brønsted
Come illustrato dalle precedente equazione generale, gli acidi di Brønsted forti come HCl, HBr, HI & H2SO4, rapidamente si addizionano al gruppo funzionale C=C degli alcheni a dare prodotti in cui nuovi legami covalenti sono formati con l'idrogeno e con la base coniugata dell'acido. Usando l'equazione sopra come guida, non è difficile capire i prodotti della reazione sotto tra il cicloesene e i sopracitati acidi di Brønsted.
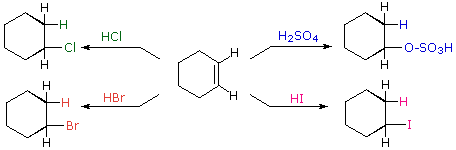 Gli acidi di Brønsted deboli come l'acqua (pKa = 15.7) e l'acido acetico (pKa = 4.75) normalmente non si addizionano agli alcheni. Comunque, l'addizione di un forte acido serve a catalizzare l'addizione di acqua, ed in questo modo possono essere preparati gli alcoli dagli alcheni. Per esempio, se l'acido solforico viene sciolto in acqua esso è completamente ionizzati a ione idronio, H3O+, e questa specie fortemente acida (pKa = -1.74) è in grado di idratare l'etilene e gli altri alcheni.
CH2=CH2 + H3O+ → CH3–CH2OH + H+
L'importanza di scegliere un solvente appropriato per queste reazioni di addizione dovrebbe essere ora chiara. Se l'addizione di HCl, HBr o HI è desiderata, non dovrebbero essere usati acqua ed alcoli. Questi acidi forti si ionizzeranno in questi solventi a dare ROH2+ e l'ossigeno nucleofilo del solvente competerà con gli anioni alogenuro nel passaggio finale, dando prodotti alcolici ed eterei. Usando solventi inerti come l'esano, il benzene e il cloruro di metilene, queste addizioni competitive del solvente sono evitate. Poiché queste reazioni procedono attraverso intermedi polari o ionici, la velocità della reazione è maggiore in solventi polari, come il nitrometano e l'acetonitrile, rispetto al caso in cui sono impiegati solventi non polari, come il cicloesano e il tetracloruro di carbonio.
Regioselettività e regola di Markovnikov
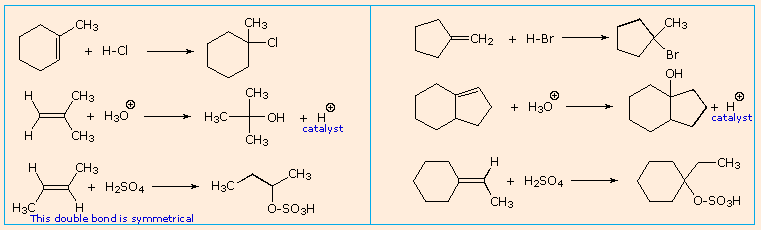
È possibile ottenere solo un prodotto dall'addizione di questi acidi forti ad alcheni simmetrici come l'etilene ed il cicloesene. Comunque, se gli atomi di carbonio del doppio legame non sono strutturalmente equivalenti, come nelle molecole dell'1-butene, del 2-metil-2-butene e dell'1-metilcicloesene, il reagente può plausibilmente addizionarsi in due modi differenti. Questo è mostrato per il 2-metil-2-butene nell'equazione seguente.
(CH3)2C=CHCH3 + H-Cl → (CH3)2CH–CHClCH3 o (CH3)2CCl–CH2CH3
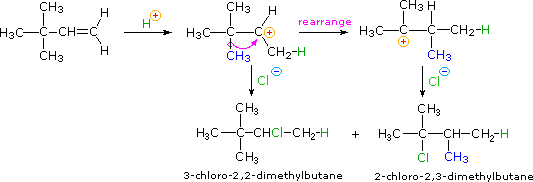
Quando le reazioni di addizione a questi alcheni non simmetrici sono portate a termine, troviamo che uno dei due possibili prodotti, isomeri costituzionali, è formato preferenzialmente. Una selettività di questo tipo è chiamata regioselettività. Nell'esempio sopra, il 2-cloro-2-metilbutano è quasi il prodotto esclusivo. In modo simile, l'1-butene forma 2-bromobutano come prodotto predominante per trattamento con HBr.
Gli acidi di Brønsted deboli come l'acqua (pKa = 15.7) e l'acido acetico (pKa = 4.75) normalmente non si addizionano agli alcheni. Comunque, l'addizione di un forte acido serve a catalizzare l'addizione di acqua, ed in questo modo possono essere preparati gli alcoli dagli alcheni. Per esempio, se l'acido solforico viene sciolto in acqua esso è completamente ionizzati a ione idronio, H3O+, e questa specie fortemente acida (pKa = -1.74) è in grado di idratare l'etilene e gli altri alcheni.
CH2=CH2 + H3O+ → CH3–CH2OH + H+
L'importanza di scegliere un solvente appropriato per queste reazioni di addizione dovrebbe essere ora chiara. Se l'addizione di HCl, HBr o HI è desiderata, non dovrebbero essere usati acqua ed alcoli. Questi acidi forti si ionizzeranno in questi solventi a dare ROH2+ e l'ossigeno nucleofilo del solvente competerà con gli anioni alogenuro nel passaggio finale, dando prodotti alcolici ed eterei. Usando solventi inerti come l'esano, il benzene e il cloruro di metilene, queste addizioni competitive del solvente sono evitate. Poiché queste reazioni procedono attraverso intermedi polari o ionici, la velocità della reazione è maggiore in solventi polari, come il nitrometano e l'acetonitrile, rispetto al caso in cui sono impiegati solventi non polari, come il cicloesano e il tetracloruro di carbonio.
Regioselettività e regola di Markovnikov
È possibile ottenere solo un prodotto dall'addizione di questi acidi forti ad alcheni simmetrici come l'etilene ed il cicloesene. Comunque, se gli atomi di carbonio del doppio legame non sono strutturalmente equivalenti, come nelle molecole dell'1-butene, del 2-metil-2-butene e dell'1-metilcicloesene, il reagente può plausibilmente addizionarsi in due modi differenti. Questo è mostrato per il 2-metil-2-butene nell'equazione seguente.
(CH3)2C=CHCH3 + H-Cl → (CH3)2CH–CHClCH3 o (CH3)2CCl–CH2CH3
Quando le reazioni di addizione a questi alcheni non simmetrici sono portate a termine, troviamo che uno dei due possibili prodotti, isomeri costituzionali, è formato preferenzialmente. Una selettività di questo tipo è chiamata regioselettività. Nell'esempio sopra, il 2-cloro-2-metilbutano è quasi il prodotto esclusivo. In modo simile, l'1-butene forma 2-bromobutano come prodotto predominante per trattamento con HBr.
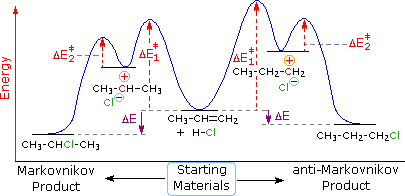
 Dopo aver studiato molte reazioni di addizione di questo tipo, il chimico russo Vladimir Markovnikov notò un trend nella struttura del prodotti di addizione favorito. Formulò questo trend come una regola empirica che ora noi chiamiamo regola di Markovnikov: Quando un acido di Brønsted, HX, si addiziona ad un doppio legame sostituito non simmetricamente, l'idrogeno acido dell'acido si lega al carbonio del doppio legame che ha il numero maggiore di atomi di idrogeno già attaccati ad esso.
Dopo aver studiato molte reazioni di addizione di questo tipo, il chimico russo Vladimir Markovnikov notò un trend nella struttura del prodotti di addizione favorito. Formulò questo trend come una regola empirica che ora noi chiamiamo regola di Markovnikov: Quando un acido di Brønsted, HX, si addiziona ad un doppio legame sostituito non simmetricamente, l'idrogeno acido dell'acido si lega al carbonio del doppio legame che ha il numero maggiore di atomi di idrogeno già attaccati ad esso.
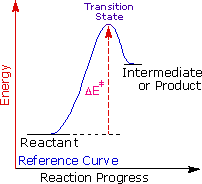
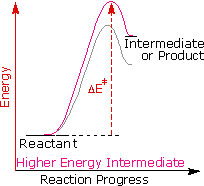
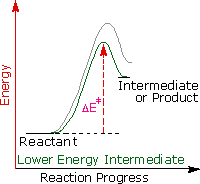
 Regole empiriche come la regola di Markovnikov sono aiuti utili per ricordare e predire risultati sperimentali. Inolte, le regole empiriche sono spesso il primo passo attraverso la padronanza di una materia, ma esse di rado costituiscono la vera comprensione. La regola di Markovnikov, per esempio, suggerisce che ci sono principi comuni ed importanti al lavoro in queste reazioni di addizione, ma essa non ci dice quali sono. Il passaggio successivo nella realizzazione e comprensione di questa reazione deve essere il costruire un modello meccanicistico razionale che possa essere testato tramite esperimento.
Tutti i reagenti discussi qui sono forti acidi di Brønsted così, come primo passaggio, sembra ragionevole trovare una base con cui l'acido possa reagire. Dato che sappiamo che questi acidi non reagiscono con gli alcani, devono essere gli elettroni π del doppio legame dell'alchene che servono come base.
Regole empiriche come la regola di Markovnikov sono aiuti utili per ricordare e predire risultati sperimentali. Inolte, le regole empiriche sono spesso il primo passo attraverso la padronanza di una materia, ma esse di rado costituiscono la vera comprensione. La regola di Markovnikov, per esempio, suggerisce che ci sono principi comuni ed importanti al lavoro in queste reazioni di addizione, ma essa non ci dice quali sono. Il passaggio successivo nella realizzazione e comprensione di questa reazione deve essere il costruire un modello meccanicistico razionale che possa essere testato tramite esperimento.
Tutti i reagenti discussi qui sono forti acidi di Brønsted così, come primo passaggio, sembra ragionevole trovare una base con cui l'acido possa reagire. Dato che sappiamo che questi acidi non reagiscono con gli alcani, devono essere gli elettroni π del doppio legame dell'alchene che servono come base.
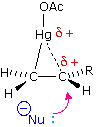
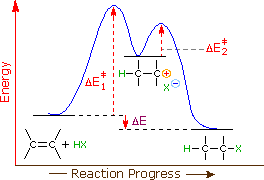
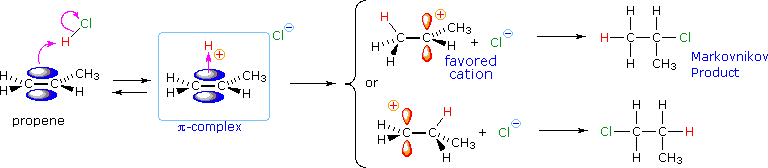
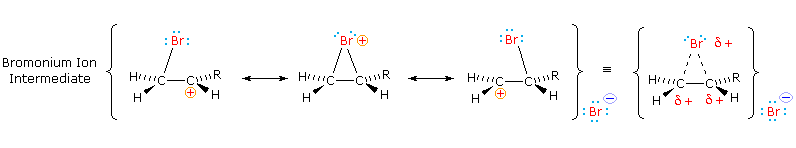
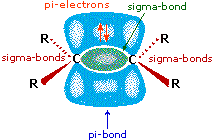 Come mostrato nell'immagine sopra, l'orbitale π si estende nello spazio immediatamente sopra e sotto il piano del doppio legame, e gli elettroni che occupano questo orbitale possono attratti verso il protone di un acido di Brønsted. Il risultante equilibrio acido-base genera un carbocatione intermedio (l'acido coniugato dell'alchene) che si combina quindi rapidamente con la base anionica coniugata dell'acido di Brønsted. Questo meccanismo a due passaggi è illustrato per la reazione dell'etilene con HCl attraverso le seguenti equazioni.
1° passaggio: H2C=CH2 + HCl → H3C–CH2+ + Cl–
2° passaggio: H3C–CH2+ + Cl– → H3C–CH2Cl
Come mostrato nell'immagine sopra, l'orbitale π si estende nello spazio immediatamente sopra e sotto il piano del doppio legame, e gli elettroni che occupano questo orbitale possono attratti verso il protone di un acido di Brønsted. Il risultante equilibrio acido-base genera un carbocatione intermedio (l'acido coniugato dell'alchene) che si combina quindi rapidamente con la base anionica coniugata dell'acido di Brønsted. Questo meccanismo a due passaggi è illustrato per la reazione dell'etilene con HCl attraverso le seguenti equazioni.
1° passaggio: H2C=CH2 + HCl → H3C–CH2+ + Cl–
2° passaggio: H3C–CH2+ + Cl– → H3C–CH2Cl
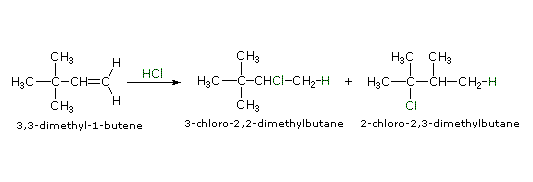
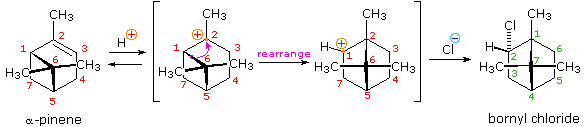
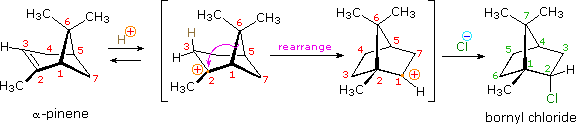
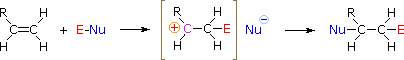
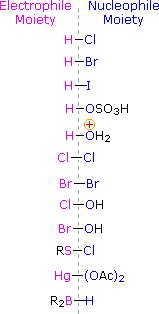
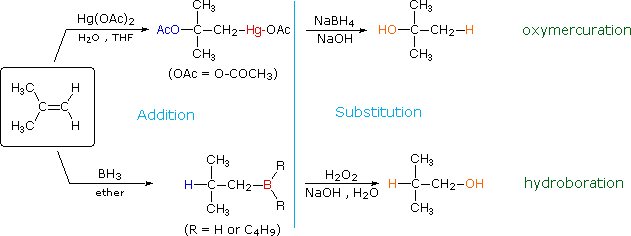
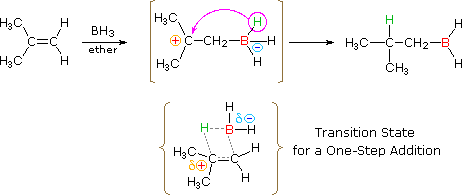
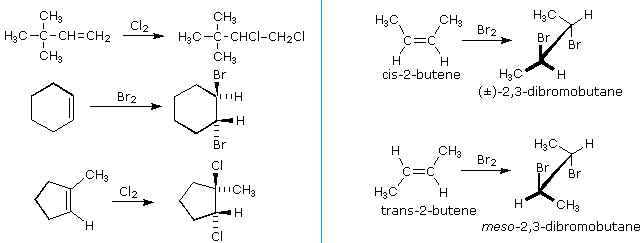
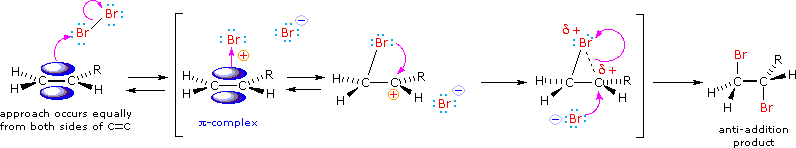
 HOCl + HCl
HOCl + HCl