quimico
2011-11-22 15:19
1. Introduzione
Il potenziale di una pandemia influenzale è una preoccupazione mondiale. Ci furono tre storiche pandemie influenzali o epidemie nel ventesimo secolo (la Spagnola nel 1918, l'Asiatica nel 1957, e quella di Hong Kong nel 1968). Un pandemia è attribuita ad una mutazione delle proteine del virus dell'influenza che permette al virus di sfuggire al sistema immunitario umano.
Il virus dell'influenza aviaria H5N1, che è nato nei volatili ad Hong Kong nel 1997, ha tali caratteristiche. Il tasso di mortalità dell'influenza aviaria è superiore al 50%. Fortunatamente, il virus al momento (era il 2008 quando uscì questo articolo) non si diffondeva da umano ad umano, sebbene ci siano paure che esso potesse presto ottenere la'abilità infettiva di fare ciò. A causa del vasto trasporto globale, un'epidemia influenzale locale non può essere limitata ad un'area specifica. Quindi, il numero dei pazienti potrebbe aumentare in modo esplosivo in diversi posti remoti simultaneamente.
A causa dell'elevata frequenza di mutazione del virus dell'influenza, un farmaco efficace anti-influenzale dovrebbe mirare ai fondamentali processi molecolari che sono essenziali e specifici per il ciclo vitale del virus. Questo principio è basato sull'ipotesi che le strutture delle proteine fondamentali (o i loro domini) vengano conservate anche in virus mutanti.
La neuraminidasi (NA) appartiene a queste proteine virali. Un virione influenzale che nasce da una cellula infetta si lega ad un residuo terminale dell'acido sialico (1) sulla superficie glicoproteica della cellula ospite con l'emagglutinina (HA: Figura 1). La NA idroliticamente rompe il legame glicosidico dell'acido sialico per rilasciare il virus dalla superficie della cellula ospite. Questo processo liberaa il virione nascente dalla cellula infettata ed è essenziale per il diffondersi dell'infezione. Come previsto, il sito attivo della NA è mantenuto strettamente attraverso la tensione del virus dell'influenza A e B.
Quindi, un inibitore della NA è un candidato primario per farmaci anti-influenzali ad ampio spettro.
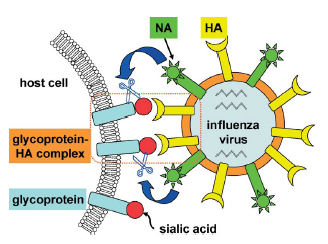
Figura 1. Rappresentazione schematica di un virione influenzale nascente da una cellula ospite.
La struttura ai raggi X del complesso NA–acido sialico (1)[1] fornisce un'informazione strutturale a livello molecolare per la progettazione di inibitori della NA. Sebbene la conformazione stabile dell'anello piranico centrale di 1 in acqua sia la forma a sedia con il suo acido carbossilico in C-1 in posizione assiale, esso si converte nella conformazione a barca 3 quando si lega ad un sito attivo della NA (Schema 1). La conformazione a barca è stabilizzata da interazioni acido–base tra l'acido carbossilico in C-1 di 1 (o 2) ed i tre residui arginina della NA al sito attivo. Questo cambiamento conformazionale è richiesto stereoelettronicamente per la rimozione del legame glicosidico. Nel conformero a barca 3, il doppieto libero dell'atomo di ossigeno del pirano è posizionato anti-parallelo rispetto al legame C–O glicosidico.
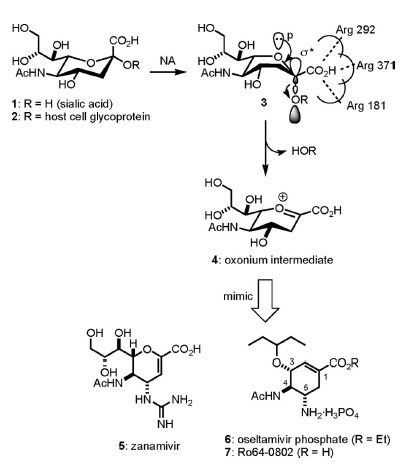
Schema 1. Uno sguardo più da vicino allo step di idrolisi dell'acido sialico attraverso la NA e la progettazione di inibitori della NA.
Il legame glicosidico è rimosso attraverso una sovrapposizione orbitalica p–σ*, ed un intermedio ossonio 4 viene generato.
Due farmaci anti-influenzali – zanamivir (5: GlaxoSmithKlein’s Relenza)[2] e oseltamivir fosfato (6: Gilead’s Tamiflu, commercializzato dalla Roche)[3] – inibiscono la NA legandosi fortemente al sito attivo della NA come analoghi stabili di 4. I valori di IC50 di questi farmaci sono nel range della nM.
L'oseltamivir fosfato è un profarmaco oralmente attivo, e la sua forma attiva è il corrispondente acido carbossilico 7.
Lo zanamivir ha minore biodisponibilità ed è somministrato per
inalazione.
Questi farmaci sono considerati essere efficaci per il trattamento dell'influenza H5N1. In accordo con l'OMS, lo stoccaggio di questi farmaci è al momento l'unica via per proteggersi contro una pandemia.
Sebbene queste molecole siano relativamente piccole, lo sviluppo di una sintesi pratica che può fornire la quantità richiesta a livello mondiale è estremamente impegnativa. Questi composti offrono quindi una opportunità urgente ma interessante per sviluppare una sintesi ideale.
Qui, sono esaminate diverte strategie sintetiche riportate in letteratura per l'oseltamivir fosfato.
2. Sintesi della Gilead
Il gruppo di medicinal chemistry alla Gilead Sciences ha sintetizzato per la prima volta il composto 6 da un prodotto naturale, l'acido (–)-shikimico, quale materiale di partenza (Schema 2)[3a].
Il trattamento del (–)-metil shikimato (8) nelle condizioni di Mitsunobu ha prodotto l'epossido tramite attivazione selettiva del gruppo idrossile meno ingombrato stericamente in C-5. Dopo protezione del rimanente gruppo idrossile in C-3 come MOM etere (9), l'apertura dell'anello epossidico con azide è avvenuta selettivamente sul meno ingombrato stericamente C-5, portando all'ottenimento dell'azido alcole 10. L'O-mesilazione, seguita da una reazione di Staudinger per generare l'imminofosforano, dalla formazione dello ione aziridinio attraverso sostituzione del gruppo mesile, e dall'idrolisi dell'intermedio con Et3N in H2O, ha prodotto l'aziridina 11. L'apertura regioselettiva dell'aziridina con azide avviene sul C-5, e la rimozione del gruppo MOM ha portato all'amino alcole 12.
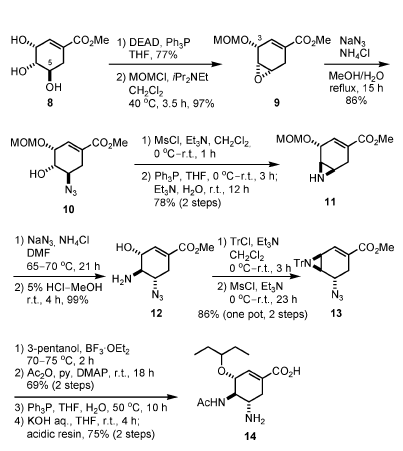
Schema 2.
L'aziridina 13, che è un precursore per l'introduzione del gruppo 3-pentilossi in C-3, è stata sintetizzata da 12 tramite un processo in due passaggi, one-pot: (1) protezione della funzionalità amino con un gruppo tritile, e (2) mesilazione del gruppo idrossi. L'apertura sito-selettiva dell'aziridina 13 con il pentan-3-olo è avvenuta in presenza di 1.5 equiv. di BF3·OEt2, e la successiva acetilazione dell'amina risultante ha prodotto il corrispondente ammido etere.
La sintesi della forma biologicamente attiva 14 dell'oseltamivir è stata completata attraverso riduzione dell'azide, seguita da idrolisi del metil estere in condizioni basiche.
Le caratteristiche di queste sintesi sono come segue: (1) l'ingombrante gruppo 3-pentilossi in posizione C-3 è introdotto nell'ultimo passaggio tramite una reazione di apertura dell'aziridina, e (2) la metà trans 1,2-diamina è costruita attraverso una reazione di apertura dell'aziridina con azide. L'introduzione nell'ultimo stadio del gruppo alcossi in C-3 è vantaggioso nello stadio di scoperta di un farmaco, perché questa parte può essere facilmente diversificata.
Anzi, il gruppo 3-pentilossi è essenziale per la potente attività inibitoria della NA[4]. Il metodo per la costruzione dell'1,2-diamina è anche utilizzato nel processo di sintesi della Roche dell'oseltamivir fosfato, discusso nella sezione successiva.
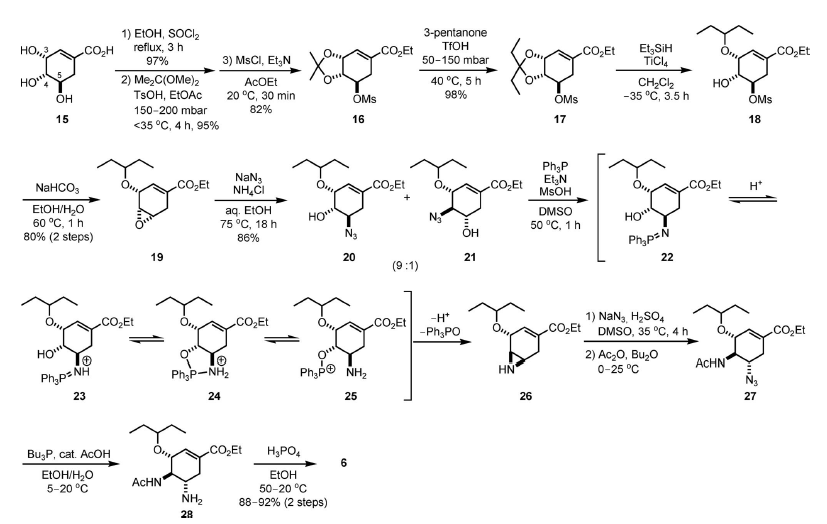 Schema 3.
L'intermedio 17 è stato sintetizzat da 15 indirettamente tramite l'acetonide cristallina 16, a causa del fatto che la purezza del composto di partenza 15 era variabile (85-99%) con profili di impurezze molto differenti. Un successivo intermedio chiave 19 non è stato ottenuto con sufficiente qualità tramite una via diretta che coinvolgeva il corrispondente pentilidene acetale. La formazione dell'etil estere sul 15, seguita da formazione dell'acetonide sui gruppi diidrossi C-3,4-cis e la mesilazione del rimanente OH in C-5, ha prodotto 16 con rese elevate. Questo composto è stato purificato tramite ricristallizzazione da MeOH. Quindi, lo scambio di acetale in presenza di un eccesso di pentan-3-one ed una quantità catalitica (4.5 mol-%) di acido triflico ha portato all'ottenimento di 17. L'apertura riduttiva regioselettiva dell'acetale è stata meglio condotta con Et3SiH e TiCl4 in CH2Cl2 a –35 °C. In queste condizioni, 18 è stata ottenuto prevalentemente (32:1) con una resa elevata, ed è stato convertio nell'epossido 19 in condizioni basiche.
La cristallizzazione da esano ha dato il composto 19 puro con una resa dell'80%.
Una reazione di apertura d'epossido con NaN3 ha prodotto una miscela 9:1 di azido alcoli isomerici 20 e 21, ed entrambi sono stati convertiti nella stessa aziridina 26. A causa della natura esplosiva degli azido composti, i composti 20 e 21 dovrebbero essere maneggiati al di sotto dei 40 °C quando puri e al di sotto dei 70 °C in soluzione.
La formazione dell'aziridina da azido alcoli comprende tre passaggi: reazione di Staudinger per ottenere l'imminofosforano 22, migrazione del trifenilfosfonio all'atomo di ossigeno in C-4 a dare 25, ed attacco SN2 all'atomo di azoto per eliminare il fosfanossido (intermedi dal 20 sono mostrati nello Schema 3 per semplificazione). Questo processo è stato facilitato in modo significativo in presenza di Et3N e MsOH, probabilmente perché la formazione della ossazafosfolidina (24) è accelerata dalla protonazione dell'atomo di azoto del 22.
NaN3 e H2SO4 sono stati aggiunti alla miscela di reazione contenente l'aziridina 26, con la temperatura mantenuta al di sotto dei 37 °C, per ottenere la corrispondente azido amina in un solo colpo da 20 e 21. Dopo N-acetilazione, è stato ottenuto 27. La riduzione di Staudinger dell'azide con Bu3P seguita da formazione del sale ha prodotto l'oseltamivir fosfato 6, che è stato purificato ad una qualità ≥99% tramite ricristallizzazione da EtOH.
Sebbene questa via sintetica possa produrre l'oseltamivir
fosfato in quantità enorme (tonnellate), c'erano due potenziali inconvenienti: (1) l'uso di intermedi contenenti azide potenzialmente esplosivi, e (2) la mancanza di disponibilità dell'acido shikimico di purezza elevata. I chimici della Roche stanno continuando la loro ricerca nei confronti dello sviluppo di vie sintetiche più sicure ed efficientis. Tre vie sintetiche alternative che affrontano questi inconvenienti sono mostrati negli Schemi 4, 5, e 6.
Per evitare l'uso dell'azide, è stata usata tBuNH2 quale alternativa fonte di amina (Schema 4)[7]. L'epossido 19 è stato aperto regioselettivamente per trattamento con il complesso tBuNH2–MgCl2, producendo l'amino alcole 29 con una resa del 96%. Grazie alla presenza di un ingombrante gruppo tert-butile sull'atomo di azoto, la mesilazione è avvenuta selettivamente sull'atomo di ossigeno, e l'aziridina 30 è stata formata in un solo colpo da 29.
Dopo apertura dell'aziridina con diallilamina in presenza di PhSO3H, l'amina secondaria è stata acetilata in condizioni relativamente forzate a dare 32, che è stato purificato tramite precipitazione come corrispondete sale di HCl. Questa operazione è l'unica purificazione richiesta nella sequenza tra 19 e 6. La rimozione del gruppo N-tert-butile in condizioni acide (TFA) e la deallilazione attraverso transfer di allile Pd-catalizzato ad acido 1,3-dimetilbarbiturico seguita da formazione del sale di fosfato ha portato al composto 6.
Schema 3.
L'intermedio 17 è stato sintetizzat da 15 indirettamente tramite l'acetonide cristallina 16, a causa del fatto che la purezza del composto di partenza 15 era variabile (85-99%) con profili di impurezze molto differenti. Un successivo intermedio chiave 19 non è stato ottenuto con sufficiente qualità tramite una via diretta che coinvolgeva il corrispondente pentilidene acetale. La formazione dell'etil estere sul 15, seguita da formazione dell'acetonide sui gruppi diidrossi C-3,4-cis e la mesilazione del rimanente OH in C-5, ha prodotto 16 con rese elevate. Questo composto è stato purificato tramite ricristallizzazione da MeOH. Quindi, lo scambio di acetale in presenza di un eccesso di pentan-3-one ed una quantità catalitica (4.5 mol-%) di acido triflico ha portato all'ottenimento di 17. L'apertura riduttiva regioselettiva dell'acetale è stata meglio condotta con Et3SiH e TiCl4 in CH2Cl2 a –35 °C. In queste condizioni, 18 è stata ottenuto prevalentemente (32:1) con una resa elevata, ed è stato convertio nell'epossido 19 in condizioni basiche.
La cristallizzazione da esano ha dato il composto 19 puro con una resa dell'80%.
Una reazione di apertura d'epossido con NaN3 ha prodotto una miscela 9:1 di azido alcoli isomerici 20 e 21, ed entrambi sono stati convertiti nella stessa aziridina 26. A causa della natura esplosiva degli azido composti, i composti 20 e 21 dovrebbero essere maneggiati al di sotto dei 40 °C quando puri e al di sotto dei 70 °C in soluzione.
La formazione dell'aziridina da azido alcoli comprende tre passaggi: reazione di Staudinger per ottenere l'imminofosforano 22, migrazione del trifenilfosfonio all'atomo di ossigeno in C-4 a dare 25, ed attacco SN2 all'atomo di azoto per eliminare il fosfanossido (intermedi dal 20 sono mostrati nello Schema 3 per semplificazione). Questo processo è stato facilitato in modo significativo in presenza di Et3N e MsOH, probabilmente perché la formazione della ossazafosfolidina (24) è accelerata dalla protonazione dell'atomo di azoto del 22.
NaN3 e H2SO4 sono stati aggiunti alla miscela di reazione contenente l'aziridina 26, con la temperatura mantenuta al di sotto dei 37 °C, per ottenere la corrispondente azido amina in un solo colpo da 20 e 21. Dopo N-acetilazione, è stato ottenuto 27. La riduzione di Staudinger dell'azide con Bu3P seguita da formazione del sale ha prodotto l'oseltamivir fosfato 6, che è stato purificato ad una qualità ≥99% tramite ricristallizzazione da EtOH.
Sebbene questa via sintetica possa produrre l'oseltamivir
fosfato in quantità enorme (tonnellate), c'erano due potenziali inconvenienti: (1) l'uso di intermedi contenenti azide potenzialmente esplosivi, e (2) la mancanza di disponibilità dell'acido shikimico di purezza elevata. I chimici della Roche stanno continuando la loro ricerca nei confronti dello sviluppo di vie sintetiche più sicure ed efficientis. Tre vie sintetiche alternative che affrontano questi inconvenienti sono mostrati negli Schemi 4, 5, e 6.
Per evitare l'uso dell'azide, è stata usata tBuNH2 quale alternativa fonte di amina (Schema 4)[7]. L'epossido 19 è stato aperto regioselettivamente per trattamento con il complesso tBuNH2–MgCl2, producendo l'amino alcole 29 con una resa del 96%. Grazie alla presenza di un ingombrante gruppo tert-butile sull'atomo di azoto, la mesilazione è avvenuta selettivamente sull'atomo di ossigeno, e l'aziridina 30 è stata formata in un solo colpo da 29.
Dopo apertura dell'aziridina con diallilamina in presenza di PhSO3H, l'amina secondaria è stata acetilata in condizioni relativamente forzate a dare 32, che è stato purificato tramite precipitazione come corrispondete sale di HCl. Questa operazione è l'unica purificazione richiesta nella sequenza tra 19 e 6. La rimozione del gruppo N-tert-butile in condizioni acide (TFA) e la deallilazione attraverso transfer di allile Pd-catalizzato ad acido 1,3-dimetilbarbiturico seguita da formazione del sale di fosfato ha portato al composto 6.
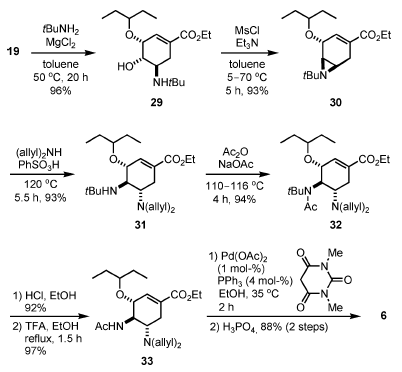 Schema 4. Sintesi azide-free dell'oseltamivir fosfato.
A causa dell'iniziale incertezza riguardo alla disponibilità di acido shikimic su scala delle tonnellate, diverse differenti strategie independenti da questo materiale di partenza furono investigate dal gruppo della Roche[6c,6d]. In modo specifico, variazioni delle strategia di Diels–Alder furono tentate. Una via efficace in questa direzione è mostrata nello Scheme 5[6c,6d].
Questa sintesi è iniziata con una reazione racema di Diels–Alder tra il furano e l'etil acrilato. La reazione avveniva in presenza di 1 equiv. di ZnCl2 senza solvente, ed è stato ottenuto il termodinamicamente più stabile eso-34 quale prodotto maggioritario (9:1). La risoluzione enzimatica del 34 ha permesso di ottenere l'isomero desiderato R 35 con un ee del 97% e conversione del 75% (20% di resa). Il composto 35 è stato convertito nella endo aziridina 38 attraverso trattamento con difenilfosforil azide (DPPA)[8] a 70 °C attraverso una cicloaddizione [3+2], portando ad una miscela di exo-36 e 37, che è stata soggetta ad estrusione termica con inversione di N2. Non è chiaro, al momento, nessun preciso meccanismo stereochimico per questa formazione di azidirina senza precedenti.
Dopo transesterificazione alla funzionalità fosfato, il trattamento con base per aprire il sistema biciclico seguito da O-mesilazione e l'apertura dell'aziridina con pentan-3-olo ha prodotto 39. L'idrolisi della fosforil ammide e la formazione del cloridrato ha portat al 40, che può essere convertito nel 6 tramite formazione di aziridina e introduzione di una funzionalità aminica in C-5, etc.
Schema 4. Sintesi azide-free dell'oseltamivir fosfato.
A causa dell'iniziale incertezza riguardo alla disponibilità di acido shikimic su scala delle tonnellate, diverse differenti strategie independenti da questo materiale di partenza furono investigate dal gruppo della Roche[6c,6d]. In modo specifico, variazioni delle strategia di Diels–Alder furono tentate. Una via efficace in questa direzione è mostrata nello Scheme 5[6c,6d].
Questa sintesi è iniziata con una reazione racema di Diels–Alder tra il furano e l'etil acrilato. La reazione avveniva in presenza di 1 equiv. di ZnCl2 senza solvente, ed è stato ottenuto il termodinamicamente più stabile eso-34 quale prodotto maggioritario (9:1). La risoluzione enzimatica del 34 ha permesso di ottenere l'isomero desiderato R 35 con un ee del 97% e conversione del 75% (20% di resa). Il composto 35 è stato convertito nella endo aziridina 38 attraverso trattamento con difenilfosforil azide (DPPA)[8] a 70 °C attraverso una cicloaddizione [3+2], portando ad una miscela di exo-36 e 37, che è stata soggetta ad estrusione termica con inversione di N2. Non è chiaro, al momento, nessun preciso meccanismo stereochimico per questa formazione di azidirina senza precedenti.
Dopo transesterificazione alla funzionalità fosfato, il trattamento con base per aprire il sistema biciclico seguito da O-mesilazione e l'apertura dell'aziridina con pentan-3-olo ha prodotto 39. L'idrolisi della fosforil ammide e la formazione del cloridrato ha portat al 40, che può essere convertito nel 6 tramite formazione di aziridina e introduzione di una funzionalità aminica in C-5, etc.
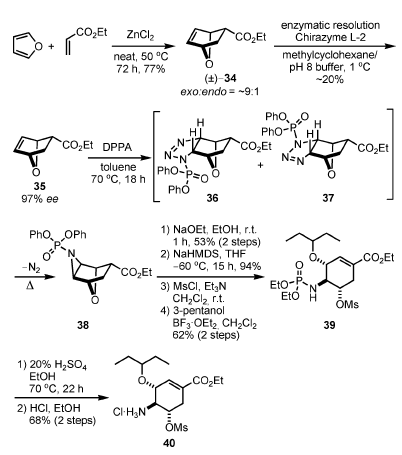 Schema 5. Sintesi dell'oseltamivir fosfato senza usare l'acido shikimico
quale materiale di partenza - strategia tipo Diels–Alder.
Una via di sintesi più promettente, anch'essa senza usare acido shikimico, è mostrata nello Schema 6[6c,6d]. Partendo dall'economico
dimetossifenolo 41, l'O-alchilazione e la successiva dibromurazione ed etossicarbonilazione hanno fornito il simmetrico 42, che è stato idrogenato su Ru/Al2O3 a dare il meso-diestere 43. Dopo selettiva O-demetilazione con TMSI, il risultante 44 è stato idrolizzato enzimaticamente con la esterasi estratta dal fegato suino (PLE), portando al monoacido 45 enantiomericamente arricchito con elevata enantioselettività (96–98% ee) e con una resa quasi quantitativa. Il riarrangiamento di Curtius del 45 per trattamento con DPPA ha introdotto un azoto in C-5 come carbammato ciclico 46, che è stato protetto con un gruppo Boc. Il risultante N-Boc-ossazolidinone è stato trattato con una quantità catalitica di NaH in toluene a riflusso, portando al corrispondente estere Δ(1,2)-α,β-insaturo. Questa formale disidratazione è avvenuta attraverso attacco intramolecolare dell'idrossido in C-2 al carbonio carbonilico dell'ossazolidinone Boc-attivato, producendo il carbonato ciclico intermedio, seguita da β-eliminazione. Il risultante gruppo idrossi in C-4 è stato quindi attivato come triflato, dando 47. L'attacco SN2 di NaN3 ha prodotto l'azide 48, che è stata ridotta con cobalto Raney, seguita da acetilazione, rimozione del Boc con HBr/AcOH, e la formazione del sale di fosfato ha prodotto 6.
Schema 5. Sintesi dell'oseltamivir fosfato senza usare l'acido shikimico
quale materiale di partenza - strategia tipo Diels–Alder.
Una via di sintesi più promettente, anch'essa senza usare acido shikimico, è mostrata nello Schema 6[6c,6d]. Partendo dall'economico
dimetossifenolo 41, l'O-alchilazione e la successiva dibromurazione ed etossicarbonilazione hanno fornito il simmetrico 42, che è stato idrogenato su Ru/Al2O3 a dare il meso-diestere 43. Dopo selettiva O-demetilazione con TMSI, il risultante 44 è stato idrolizzato enzimaticamente con la esterasi estratta dal fegato suino (PLE), portando al monoacido 45 enantiomericamente arricchito con elevata enantioselettività (96–98% ee) e con una resa quasi quantitativa. Il riarrangiamento di Curtius del 45 per trattamento con DPPA ha introdotto un azoto in C-5 come carbammato ciclico 46, che è stato protetto con un gruppo Boc. Il risultante N-Boc-ossazolidinone è stato trattato con una quantità catalitica di NaH in toluene a riflusso, portando al corrispondente estere Δ(1,2)-α,β-insaturo. Questa formale disidratazione è avvenuta attraverso attacco intramolecolare dell'idrossido in C-2 al carbonio carbonilico dell'ossazolidinone Boc-attivato, producendo il carbonato ciclico intermedio, seguita da β-eliminazione. Il risultante gruppo idrossi in C-4 è stato quindi attivato come triflato, dando 47. L'attacco SN2 di NaN3 ha prodotto l'azide 48, che è stata ridotta con cobalto Raney, seguita da acetilazione, rimozione del Boc con HBr/AcOH, e la formazione del sale di fosfato ha prodotto 6.
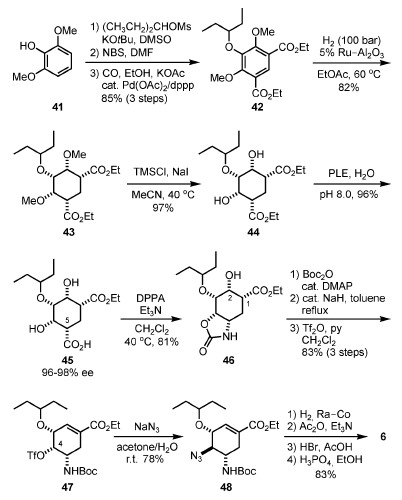 Schema 6. Sintesi dell'oseltamivir fosfato senza usare acido shikimico
quale materiale di partenza – strategia di desimmetrizzazione.
Schema 6. Sintesi dell'oseltamivir fosfato senza usare acido shikimico
quale materiale di partenza – strategia di desimmetrizzazione.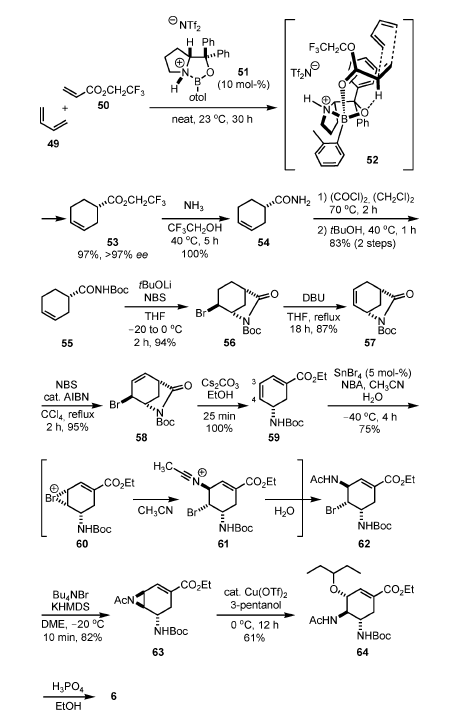
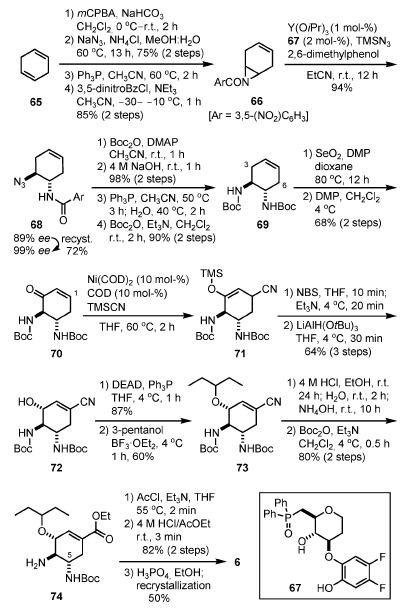 Schema 8. Sintesi di Shibasaki e Kanai di prima generazione.
Attraverso l'uso di un complesso (1 mol-%) generato da Y(OiPr)3 e il ligando chirale 67 miscelati in un rapporto 1:2 come catalizzatore asimmetrico[15], è avvenuta una reazione di apertura d'anello asimmetrica di 66 con TMSN3 a temperatura ambiente in presenza di 2,6-dimetilfenolo (1 equiv.).
Il prodotto 68 è stato ottenuto con un ee dell'89% e con una resa del 94%. La purezza enantiomerica è stata arricchita fino ad un ee del 99% ee attraverso un recristallizzazione da iPrOH.
Il catalizzatore attivo è stato determinato essere un complesso polimetallico (Y/67 = 2:3 o complesso 4:5)[16], e la reazione è proseguita attraverso un transfer intramolecolare di azide dall'azide di ittrio (generata tramite transmetallazione da TMSN3) ad un'aziridina attivata che è coordinata ad un altro ittrio che agisce da acido di Lewis (Figura 2)[17].
Schema 8. Sintesi di Shibasaki e Kanai di prima generazione.
Attraverso l'uso di un complesso (1 mol-%) generato da Y(OiPr)3 e il ligando chirale 67 miscelati in un rapporto 1:2 come catalizzatore asimmetrico[15], è avvenuta una reazione di apertura d'anello asimmetrica di 66 con TMSN3 a temperatura ambiente in presenza di 2,6-dimetilfenolo (1 equiv.).
Il prodotto 68 è stato ottenuto con un ee dell'89% e con una resa del 94%. La purezza enantiomerica è stata arricchita fino ad un ee del 99% ee attraverso un recristallizzazione da iPrOH.
Il catalizzatore attivo è stato determinato essere un complesso polimetallico (Y/67 = 2:3 o complesso 4:5)[16], e la reazione è proseguita attraverso un transfer intramolecolare di azide dall'azide di ittrio (generata tramite transmetallazione da TMSN3) ad un'aziridina attivata che è coordinata ad un altro ittrio che agisce da acido di Lewis (Figura 2)[17].
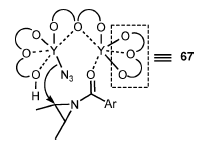 Figura 2. Rappresentazione schematica che mostra il meccanismo della reazione catalitica di apertura d'anello (aziridina) asimmetrica (Y/67 = il complesso 2:3 è mostrato come un catalizzatore rappresentativo).
Questa reazione è stata condotta sun una scala relativamente grande (30 g) senza difficoltà. Dopo la reazione, il ligando 67 è stato recuperato con una resa dell'81% tramite estrazione con una base. Il ligando recuperato può essere riusato senza alcuna diminuzione nell'attività catalitica e della enantioselettività.
La sintesi di 6 da 68 ha richiesto l'introduzione di una funzione ossigenata in C-3 e del gruppo etossicarbonile in C-1.
Per introdurre la funzionalità ossigenata in C-3, 68 è stato prima convertito nel diBoc 69 C2-simmetrico attraverso formazione della Boc immide, idrolisi del gruppo benzoile, riduzione di Staudinger, e protezione N-Boc. L'ossidazione allilica è stata condotta con SeO2[18] in presenza del periodinano di Dess–Martin (DMP). L'aggiunta del DMP ha efficacemente prevenuto la sovra-ossidazione a dare il 3,6-diolo tramite conversione del monoalcole allilico prodotto nel corrispondente enone 70 nella miscela di reazione.
Dopo completa conversione del rimanente alcole allilico nell'enone tramite ossidazione con DMP, il gruppo CN è stato introdotto in C-1 tramite il loro metodo originale con il complesso Ni–COD quale catalizzatore[19].
L'α-bromurazione del silil enol etere 71 così generato con NBS, seguita dall'eliminazione di HBr tramite uso di Et3N quale base, ha prodotto il corrispondente β-ciano enone. Questo enone non era molto stabile durante la purificazione su gel di silice, e così è stato immediatamente ridotto senza purificazione con l'agente riducente ingombrato LiAlH(OtBu)3 a dare l'alcole α-allilico 72 con eccellente stereoselettività (>20:1).
La formazione dell'aziridina nell condizioni di Mitsunobu[20] e la successiva apertura regioselettiva dell'aziridina con pentan-3-olo in posizione allilica (C-3) ha prodotto 73. L'etanolisi del nitrile è proseguita con la concomitante rimozione del Boc per trattamento con etanolo acido, e la risultante diamina è stata monoprotetta selettivamente con un gruppo Boc sull'azoto in C-5, dando 74.
L'N-acetilazione seguita da rimozione del Boc e formazione del sale di fosfato ha dato il prodotto 6[21].
La loro via sintetica di prima generazione si basava sulla reazione pratica, asimmetrica, di apertura dell'aziridina con TMSN3.
Comunque, l'uso del reagente di selenio, tossico, nell'ossidazione allilica ed il necessario trasferimento del gruppo protettivo sono stati i due maggiori inconvenienti.
Questi inconvenienti sono stati superati nella loro sintesi di seconda generazione (Schema 9)[22]. Dopo la reazione di apertira di aziridina asimmetrica, l'N-Boc azide 75 è stata sintetizzata tramite la medesima procedura come descritto nello Schema 8 (passaggi 1 e 2 della conversione da 68 a 69).
La iodolattamizzazione seguita da eliminazione di HI per trattamento con DBU ha prodotto il carbonato biciclico 76. La protezione dell'atmo di azoto del carbammato con un gruppo Boc e l'acetilazione riduttiva dell'azide con AcSH[23] ha dato 77, che è stato idrolizzato selettivamente ed ossidato con DMP per produrre l'enone 78. La cianofosforilazione del 78 con DEPC è proseguita in
presenza di LiCN[24] ed il cianofosfato 79 è stato ottenuto quale singolo isomero rilevabile. Il riarrangiamento allilico del fosfato in condizioni termiche[25] ha prodotto il corrispondente α-fosfato allilico, che è stato trattato con NH4Cl acquoso a dare il β-alcole 80 attraverso sostituzione SN2 del fosfato con H2O. Dopo inversione della stereochimica allilica tramite una reazione di Mitsunobu con acido p-nitrobenzoico, seguita da idrolisi del benzoato in condizioni basiche, l'azidirina 82 è stata sintetizzata tramite una reazione di Mitsunobu. L'apertura dell'aziridina con pentan-3-olo, la rimozione del Boc con concomitante etanolisi del nitrile, e formazione del sale di fosfato hanno prodotto 6.
Figura 2. Rappresentazione schematica che mostra il meccanismo della reazione catalitica di apertura d'anello (aziridina) asimmetrica (Y/67 = il complesso 2:3 è mostrato come un catalizzatore rappresentativo).
Questa reazione è stata condotta sun una scala relativamente grande (30 g) senza difficoltà. Dopo la reazione, il ligando 67 è stato recuperato con una resa dell'81% tramite estrazione con una base. Il ligando recuperato può essere riusato senza alcuna diminuzione nell'attività catalitica e della enantioselettività.
La sintesi di 6 da 68 ha richiesto l'introduzione di una funzione ossigenata in C-3 e del gruppo etossicarbonile in C-1.
Per introdurre la funzionalità ossigenata in C-3, 68 è stato prima convertito nel diBoc 69 C2-simmetrico attraverso formazione della Boc immide, idrolisi del gruppo benzoile, riduzione di Staudinger, e protezione N-Boc. L'ossidazione allilica è stata condotta con SeO2[18] in presenza del periodinano di Dess–Martin (DMP). L'aggiunta del DMP ha efficacemente prevenuto la sovra-ossidazione a dare il 3,6-diolo tramite conversione del monoalcole allilico prodotto nel corrispondente enone 70 nella miscela di reazione.
Dopo completa conversione del rimanente alcole allilico nell'enone tramite ossidazione con DMP, il gruppo CN è stato introdotto in C-1 tramite il loro metodo originale con il complesso Ni–COD quale catalizzatore[19].
L'α-bromurazione del silil enol etere 71 così generato con NBS, seguita dall'eliminazione di HBr tramite uso di Et3N quale base, ha prodotto il corrispondente β-ciano enone. Questo enone non era molto stabile durante la purificazione su gel di silice, e così è stato immediatamente ridotto senza purificazione con l'agente riducente ingombrato LiAlH(OtBu)3 a dare l'alcole α-allilico 72 con eccellente stereoselettività (>20:1).
La formazione dell'aziridina nell condizioni di Mitsunobu[20] e la successiva apertura regioselettiva dell'aziridina con pentan-3-olo in posizione allilica (C-3) ha prodotto 73. L'etanolisi del nitrile è proseguita con la concomitante rimozione del Boc per trattamento con etanolo acido, e la risultante diamina è stata monoprotetta selettivamente con un gruppo Boc sull'azoto in C-5, dando 74.
L'N-acetilazione seguita da rimozione del Boc e formazione del sale di fosfato ha dato il prodotto 6[21].
La loro via sintetica di prima generazione si basava sulla reazione pratica, asimmetrica, di apertura dell'aziridina con TMSN3.
Comunque, l'uso del reagente di selenio, tossico, nell'ossidazione allilica ed il necessario trasferimento del gruppo protettivo sono stati i due maggiori inconvenienti.
Questi inconvenienti sono stati superati nella loro sintesi di seconda generazione (Schema 9)[22]. Dopo la reazione di apertira di aziridina asimmetrica, l'N-Boc azide 75 è stata sintetizzata tramite la medesima procedura come descritto nello Schema 8 (passaggi 1 e 2 della conversione da 68 a 69).
La iodolattamizzazione seguita da eliminazione di HI per trattamento con DBU ha prodotto il carbonato biciclico 76. La protezione dell'atmo di azoto del carbammato con un gruppo Boc e l'acetilazione riduttiva dell'azide con AcSH[23] ha dato 77, che è stato idrolizzato selettivamente ed ossidato con DMP per produrre l'enone 78. La cianofosforilazione del 78 con DEPC è proseguita in
presenza di LiCN[24] ed il cianofosfato 79 è stato ottenuto quale singolo isomero rilevabile. Il riarrangiamento allilico del fosfato in condizioni termiche[25] ha prodotto il corrispondente α-fosfato allilico, che è stato trattato con NH4Cl acquoso a dare il β-alcole 80 attraverso sostituzione SN2 del fosfato con H2O. Dopo inversione della stereochimica allilica tramite una reazione di Mitsunobu con acido p-nitrobenzoico, seguita da idrolisi del benzoato in condizioni basiche, l'azidirina 82 è stata sintetizzata tramite una reazione di Mitsunobu. L'apertura dell'aziridina con pentan-3-olo, la rimozione del Boc con concomitante etanolisi del nitrile, e formazione del sale di fosfato hanno prodotto 6.
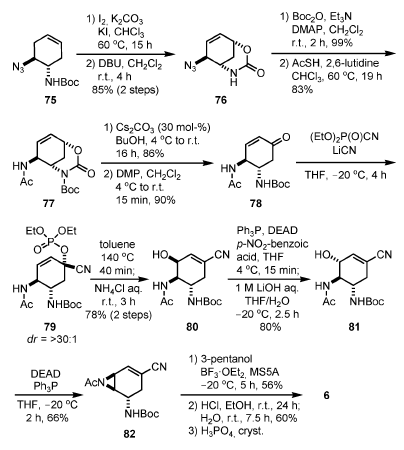 Schema 9. Sintesi di Shibasaki e Kanai di seconda generazione.
Sebbene i due inconvenienti della sintesi di prima generazione fossero stati eliminati, la via sintetica di seconda generazione era ancora lunga (20 passaggi da 65). Nel 2007, gli autori svilupparono una via sintetica completamente differente usando la reazione di Diels–Alder
ed il riarrangiamento di Curtius quali passaggi chiave (Schema 10)[26]. Sebbene la via sintetica di terza generazione dipendesse dalla risoluzione tramite HPLC chirale, essa richiedeva significativamente meno passaggi (12 passaggi) rispetto alle sintesi di prima e seconda generazione.
Schema 9. Sintesi di Shibasaki e Kanai di seconda generazione.
Sebbene i due inconvenienti della sintesi di prima generazione fossero stati eliminati, la via sintetica di seconda generazione era ancora lunga (20 passaggi da 65). Nel 2007, gli autori svilupparono una via sintetica completamente differente usando la reazione di Diels–Alder
ed il riarrangiamento di Curtius quali passaggi chiave (Schema 10)[26]. Sebbene la via sintetica di terza generazione dipendesse dalla risoluzione tramite HPLC chirale, essa richiedeva significativamente meno passaggi (12 passaggi) rispetto alle sintesi di prima e seconda generazione.
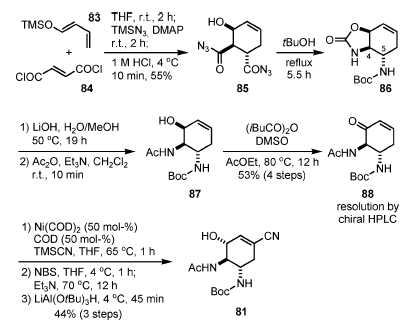 Schema 10. Sintesi di Shibasaki e Kanai di terza generazione.
La reazione di Diels–Alder tra il silossi diene 83 ed il fumaroil cloruro (84) è avvenuta a temperatura ambiente in 2 h. Dopo completamento della reazione, TMSN3 e DMAP sono stati aggiunti alla miscela, ed è stata formata la corrispondente acil azide.
Tranute quenching della reazione con HCl (1M), è avvenuta la desililazione ed è stato ottenuto 85 con una resa del 55% su tre operazioni.
Sebbene la reazione di Diels–Alder abbia dato una miscela 2:1 (endo/eso) di diastereoisomeri, l'indesiderato isomero eso è stato selettivamente decomposto durante la rimozione acida del TMS etere. Un riarrangiamento di Curtius è stato condotto tramite riscaldamento di una soluzione in tBuOH dell'acil azide 85 a riflusso.
In questo processo, i due atomi di azoto in at C-4 e C-5 sono stati differenziati, portando a 86. L'idrolisi selettiva del carbonato ciclico con LiOH e la successiva N-acetilazione ha prodotto 87, che è stato ossidato all'enone 88 nelle condizioni di Moffat (modificate), con anidride isobutirrica[27].
L'suo della stericamente ingombrata anidride in questo passaggio è stato essenziale al fine si evitare l'O-acilazione. A questo stadio, gli enantiomeri sono stati separati tramite HPLC chirale.
L'addizione coniugata mediata da Ni di TMSCN, seguita da α-bromurazione, eliminazione di HBr, e riduzione stereoselettiva LiAl(OtBu)3H, ha portato all'ottenimento di 81, il medesimo intermedio come nell loro sintesi di seconda generazione.
L'estensione di questa via di sintesi ad una variante asimmetrica tramite lo sviluppo di una reazione asimmetrica, catalitica, di Diels–Alder era, nel 2008, in corso.
Schema 10. Sintesi di Shibasaki e Kanai di terza generazione.
La reazione di Diels–Alder tra il silossi diene 83 ed il fumaroil cloruro (84) è avvenuta a temperatura ambiente in 2 h. Dopo completamento della reazione, TMSN3 e DMAP sono stati aggiunti alla miscela, ed è stata formata la corrispondente acil azide.
Tranute quenching della reazione con HCl (1M), è avvenuta la desililazione ed è stato ottenuto 85 con una resa del 55% su tre operazioni.
Sebbene la reazione di Diels–Alder abbia dato una miscela 2:1 (endo/eso) di diastereoisomeri, l'indesiderato isomero eso è stato selettivamente decomposto durante la rimozione acida del TMS etere. Un riarrangiamento di Curtius è stato condotto tramite riscaldamento di una soluzione in tBuOH dell'acil azide 85 a riflusso.
In questo processo, i due atomi di azoto in at C-4 e C-5 sono stati differenziati, portando a 86. L'idrolisi selettiva del carbonato ciclico con LiOH e la successiva N-acetilazione ha prodotto 87, che è stato ossidato all'enone 88 nelle condizioni di Moffat (modificate), con anidride isobutirrica[27].
L'suo della stericamente ingombrata anidride in questo passaggio è stato essenziale al fine si evitare l'O-acilazione. A questo stadio, gli enantiomeri sono stati separati tramite HPLC chirale.
L'addizione coniugata mediata da Ni di TMSCN, seguita da α-bromurazione, eliminazione di HBr, e riduzione stereoselettiva LiAl(OtBu)3H, ha portato all'ottenimento di 81, il medesimo intermedio come nell loro sintesi di seconda generazione.
L'estensione di questa via di sintesi ad una variante asimmetrica tramite lo sviluppo di una reazione asimmetrica, catalitica, di Diels–Alder era, nel 2008, in corso. Schema 11. Protocollo per la sintesi di un oseltamivir da usare come sonda per la PET.
Sebbene questo studio utilizzasse del normale (contenente 12C) AcCl, può essere esteso alla sintesi di un tracciante radioattivo per PET usando CH311COCl. Studi per chiarire la distribuzione dell'oseltamivir con l'aiuto della PET erano in corso.
Schema 11. Protocollo per la sintesi di un oseltamivir da usare come sonda per la PET.
Sebbene questo studio utilizzasse del normale (contenente 12C) AcCl, può essere esteso alla sintesi di un tracciante radioattivo per PET usando CH311COCl. Studi per chiarire la distribuzione dell'oseltamivir con l'aiuto della PET erano in corso.
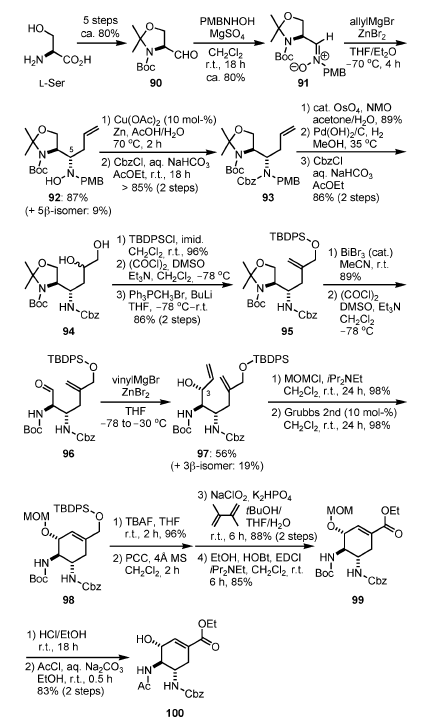 Schema 12.
La aldeide L di Garner (90), ottenuta in cinque passaggi da L-serina, è stata condensta con N-(p-metossibenzil)idrossilamina (PMBNHOH), a dare il nitrone 91. L'addizione di allilmagnesio bromuro
in presenza di una quantità stechiometrica di ZnBr2 ha prodotto la desiderata 5α-idrossilamina 92 quale maggiore isomero (87 %), forse attraverso uno stato di transizione ciclico[31]. Dopo rimozione riduttiva di idrossilamina, seguita da protezione dell'atomo di azoto con un gruppo Cbz a dare 93, diidrossilazione del doppio legame terminale, rimozione idrogenolitica del gruppo Cbz e dei gruppi PMBs, e riprotezione dell'atomo di azoto in C-5 con Cbz ha prodotto il diolo 94. La protezione selettiva del gruppo idrossi primario con un gruppo TBDPS, l'ossidazione di Swern del rimanente alcole secondario a chetone, ed una reazione di Wittig hanno portato all'olefina 95, che ha agito come braccio per l'ultimo stadio di RCM.
L'altro doppio legame C=C per la RCM è stato introdotto attraverso una rimozione dell'acetale in presenza di BiBr3 catalitico,
ossidazione di Swern, ed addizione di vinil metallo, dando 97, con la desiderata stereochimica al C-3, quale isomero maggioritario (dr = 3:1). La protezione del gruppo idrossi in C-3 con un gruppo MOM, seguita da RCM in presenza del catalizzatore di Grubbs di seconda generazione, ha prodotto 98, contenente il nucleo cicloesenico dell'oseltamivir.
Il gruppo idrossi primario, generato tramite desililazione, è stato ossidato in due passaggi con PCC e NaClO2. Il risultante acido carbossilico è stato quindi convertito nel corrispondente etil estere a dare 99.
Entrambi i gruppi MOM e Boc sono stati rimossi con etanolo acido e la risultante amina è stata acetilata a dare 100.
Sebbene la via sintetica di Yao utilizzi abbondante L-serina quale materiale di partenza, la sintesi è abbastanza lunga e la stereoinduzione del C-3 non è efficiente. Inoltre, è discutibile che 100 possa essere un precursore di 6[32].
Schema 12.
La aldeide L di Garner (90), ottenuta in cinque passaggi da L-serina, è stata condensta con N-(p-metossibenzil)idrossilamina (PMBNHOH), a dare il nitrone 91. L'addizione di allilmagnesio bromuro
in presenza di una quantità stechiometrica di ZnBr2 ha prodotto la desiderata 5α-idrossilamina 92 quale maggiore isomero (87 %), forse attraverso uno stato di transizione ciclico[31]. Dopo rimozione riduttiva di idrossilamina, seguita da protezione dell'atomo di azoto con un gruppo Cbz a dare 93, diidrossilazione del doppio legame terminale, rimozione idrogenolitica del gruppo Cbz e dei gruppi PMBs, e riprotezione dell'atomo di azoto in C-5 con Cbz ha prodotto il diolo 94. La protezione selettiva del gruppo idrossi primario con un gruppo TBDPS, l'ossidazione di Swern del rimanente alcole secondario a chetone, ed una reazione di Wittig hanno portato all'olefina 95, che ha agito come braccio per l'ultimo stadio di RCM.
L'altro doppio legame C=C per la RCM è stato introdotto attraverso una rimozione dell'acetale in presenza di BiBr3 catalitico,
ossidazione di Swern, ed addizione di vinil metallo, dando 97, con la desiderata stereochimica al C-3, quale isomero maggioritario (dr = 3:1). La protezione del gruppo idrossi in C-3 con un gruppo MOM, seguita da RCM in presenza del catalizzatore di Grubbs di seconda generazione, ha prodotto 98, contenente il nucleo cicloesenico dell'oseltamivir.
Il gruppo idrossi primario, generato tramite desililazione, è stato ossidato in due passaggi con PCC e NaClO2. Il risultante acido carbossilico è stato quindi convertito nel corrispondente etil estere a dare 99.
Entrambi i gruppi MOM e Boc sono stati rimossi con etanolo acido e la risultante amina è stata acetilata a dare 100.
Sebbene la via sintetica di Yao utilizzi abbondante L-serina quale materiale di partenza, la sintesi è abbastanza lunga e la stereoinduzione del C-3 non è efficiente. Inoltre, è discutibile che 100 possa essere un precursore di 6[32].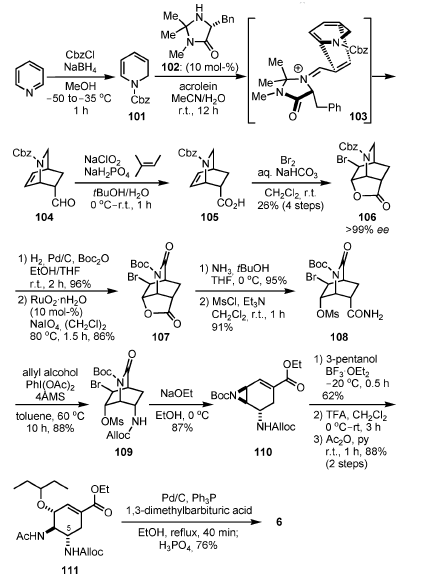 Schema 13.
La diidropiridina 101 è stata sintetizzata da piridina tramite riduzione con NaBH4 in presenza di CbzCl. Una reazione di Diels–Alder asimmetrica tra 101 e acroleina è avvenuta in presenza di 10 mol-% di 102, producendo il biciclo 104. L'ossidazione di Kraus seguita da bromolattonizzazione ha prodotto 106 con una resa del 26% dalla piridina (4 passaggi). La resa moderata è stata attribuita ad una reazione di Diels–Alder asimmetrica (<30%), ma 106 è stato facilmente purificato attraverso partizione acido/base e cristallizzazione. Il 106 chimicamente ed enantiomericamente puro è stato ottenuto senza cromatrografia su colonna. L'eccellente enantioselettività nella reazione di Diels–Alder può essere spiegata attraverso considerazione del modello 103, originalmente proposto da MacMillan: il diene 101 dovrebbe avvicinarsi al dienofilo attivato
dal lato opposto del gruppo stericamente ingombrato (gruppo benzile) rappresentato dal catalizzatore[34].
Dopo che il gruppo Cbz del 106 è stato scambiato con un gruppo Boc, l'ossidazione con una quantità catalitica di RuO2·nH2O ed una quantità stechiometrica di NaIO4 si è ottenuta l'immide 107. Il RuO2·nH2O è stato recuperato per esser riusato tramite quenching della reazione con iPrOH e filtrazione. In aggiunta, il solvente (CH2Cl)2 può essere sostituito con il più salubre dal punto di vista ambientale AcOPr senza diminuzione dell'efficienza. L'ammonolisi del lattone seguita da O-mesilazione
ha prodotto l'ammide 108, che è stata soggetta a riarrangiamento di Hofmann in presenza di PhI(OAc)2 ed alcole allilico[35] a dare l'allil carbammato 109. Il trattamento di 109 con NaOEt ha portato all'aziridina 110 attraverso etanolisi dell'immide, aziridinazione, ed eliminazione di HBr in un solo colpo.
Una reazione di apertura dell'aziridina con pentan-3-olo, rimozione del gruppo Boc con TFA, N-acetilazione, deprotezione dell'amina in C-5, e formazione del sale di fosfato ha prodotto 6.
Sebbene la resa della reazione catalitica asimmetrica di Diels–Alder richieda ulteriori miglioramenti, la sintesi di Fukuyama in 14 passaggi è entro il range pratico grazie ai seguenti punti: (1) il materiale di partenza è la piridina a buon mercato, (2) il numero di purificazioni in colonna è minimo, e (3) i reagenti innocui e comunemente usati sono usati per la sintesi.
Schema 13.
La diidropiridina 101 è stata sintetizzata da piridina tramite riduzione con NaBH4 in presenza di CbzCl. Una reazione di Diels–Alder asimmetrica tra 101 e acroleina è avvenuta in presenza di 10 mol-% di 102, producendo il biciclo 104. L'ossidazione di Kraus seguita da bromolattonizzazione ha prodotto 106 con una resa del 26% dalla piridina (4 passaggi). La resa moderata è stata attribuita ad una reazione di Diels–Alder asimmetrica (<30%), ma 106 è stato facilmente purificato attraverso partizione acido/base e cristallizzazione. Il 106 chimicamente ed enantiomericamente puro è stato ottenuto senza cromatrografia su colonna. L'eccellente enantioselettività nella reazione di Diels–Alder può essere spiegata attraverso considerazione del modello 103, originalmente proposto da MacMillan: il diene 101 dovrebbe avvicinarsi al dienofilo attivato
dal lato opposto del gruppo stericamente ingombrato (gruppo benzile) rappresentato dal catalizzatore[34].
Dopo che il gruppo Cbz del 106 è stato scambiato con un gruppo Boc, l'ossidazione con una quantità catalitica di RuO2·nH2O ed una quantità stechiometrica di NaIO4 si è ottenuta l'immide 107. Il RuO2·nH2O è stato recuperato per esser riusato tramite quenching della reazione con iPrOH e filtrazione. In aggiunta, il solvente (CH2Cl)2 può essere sostituito con il più salubre dal punto di vista ambientale AcOPr senza diminuzione dell'efficienza. L'ammonolisi del lattone seguita da O-mesilazione
ha prodotto l'ammide 108, che è stata soggetta a riarrangiamento di Hofmann in presenza di PhI(OAc)2 ed alcole allilico[35] a dare l'allil carbammato 109. Il trattamento di 109 con NaOEt ha portato all'aziridina 110 attraverso etanolisi dell'immide, aziridinazione, ed eliminazione di HBr in un solo colpo.
Una reazione di apertura dell'aziridina con pentan-3-olo, rimozione del gruppo Boc con TFA, N-acetilazione, deprotezione dell'amina in C-5, e formazione del sale di fosfato ha prodotto 6.
Sebbene la resa della reazione catalitica asimmetrica di Diels–Alder richieda ulteriori miglioramenti, la sintesi di Fukuyama in 14 passaggi è entro il range pratico grazie ai seguenti punti: (1) il materiale di partenza è la piridina a buon mercato, (2) il numero di purificazioni in colonna è minimo, e (3) i reagenti innocui e comunemente usati sono usati per la sintesi.
 Schema 14.
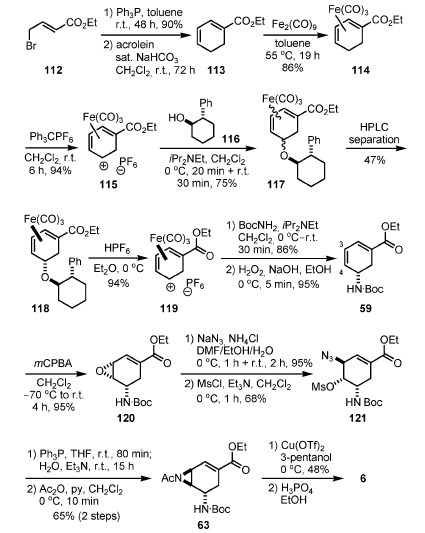
Il catione 119 è stato quindi intrappolato con BocNH2 dal lato opposto del gruppo ferro carbonile, ed una successiva decomplessazione ossidativa con perossido di idrogeno basico ha dato il dienil estere 59. L'epossidazione selettiva del più elettron-ricco doppio legame C=C tra il C-3 ed il C-4 con mCPBA ha condotto all'epossido 120. La mesil azide 121 è stata quindi sintetizzata da 120 attraverso una reazione d'apertura dell'epossido con NaN3, seguita da mesilazione. Una riduzione di Staudinger dell'azide con Ph3P e la successiva idrolisi hanno prodotto l'aziridina, che è stata acetilata producendo l'intermedio di Corey 63.
L'oseltamivir è stato quindi sintetizzato da 63 tramite la procedura di Corey.
9. Sintesi di Fang
Il gruppo di Fang sviluppò una nuova via sintetica per 6, rivolta verso una sintesi analoga (Schema 15)[38]. Specificamente, sono stati sintetizzati un analogo di un fosfonato (132: Tamiphosphor) e i composti contenenti guanidina 134 e 135.
Questi analoghi mostrano in particolare un'attività inibitoria nei confronti della neuraminidasi di maggiore potenza rispetto a 6.
Schema 14.
Il catione 119 è stato quindi intrappolato con BocNH2 dal lato opposto del gruppo ferro carbonile, ed una successiva decomplessazione ossidativa con perossido di idrogeno basico ha dato il dienil estere 59. L'epossidazione selettiva del più elettron-ricco doppio legame C=C tra il C-3 ed il C-4 con mCPBA ha condotto all'epossido 120. La mesil azide 121 è stata quindi sintetizzata da 120 attraverso una reazione d'apertura dell'epossido con NaN3, seguita da mesilazione. Una riduzione di Staudinger dell'azide con Ph3P e la successiva idrolisi hanno prodotto l'aziridina, che è stata acetilata producendo l'intermedio di Corey 63.
L'oseltamivir è stato quindi sintetizzato da 63 tramite la procedura di Corey.
9. Sintesi di Fang
Il gruppo di Fang sviluppò una nuova via sintetica per 6, rivolta verso una sintesi analoga (Schema 15)[38]. Specificamente, sono stati sintetizzati un analogo di un fosfonato (132: Tamiphosphor) e i composti contenenti guanidina 134 e 135.
Questi analoghi mostrano in particolare un'attività inibitoria nei confronti della neuraminidasi di maggiore potenza rispetto a 6.
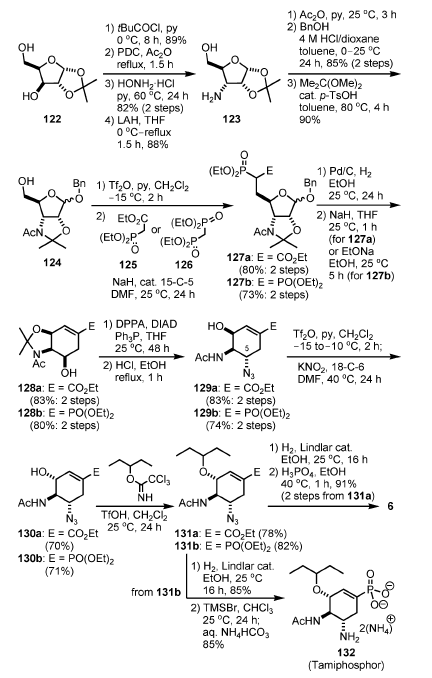 Schema 15.
La sintesi iniziava dal derivato del D-xylosio 122. Dopo protezione selettiva del gruppo idrossi primario con una funzionalità pivaloile, il gruppo idrossi secondario è stato convertito in un α-amino gruppo (123) attraverso ossidazione, formazione dell'ossima, e riduzione.
N-acetilazione, introduzione del gruppo benzilossi in posizione anomerica in condizioni acide, e formazione dell'acetale hanno prodotto 124. La parte fosfato è stata introdotto attraverso attivazione del gruppo idrossi primario come triflato, seguita da sostituzione tramite l'anione fosfonato derivato da 125 o 126, producendo 127a e 127b, rispettivamente. Il gruppo benzile anomerico è stato rimosso idrogeneticamente, ed il nucleo cicloesenico dei composti d'interesse è stato costruito tramite un reazione intramolecolare di Horner–Wadsworth–Emmons, portando al 128.
I rimanenti compiti a questo stadio erano: (1) conversione dell'OH in C-5 in una funzionalità amino, e (2) introduzione del gruppo 3-pentilossi in C-3.
La conversione dell'OH in C-5 in una funzionalità amino è stata ottenuta attraverso inversione di Mitsunobu del gruppo β-idrossi in C-5 con DPPA, diisopropil azodicarbossilato (DIAD), e Ph3P[39]. Dopo idrolisi dell'acetale in condizioni acide, il gruppo idrossi in C-3 è stato invertito attraverso formazione del triflato seguita da attacco SN2 con KNO2. Il gruppo 3-pentile è stato quindi introdotto attraverso sostituzione del 3-pentilossi immidato attraverso l'ossigeno in C-3 in presenza di TfOH. Infine, l'idrogenolisi dell'azide in presenza del catalizzatore di Lindlar e la successiva formazione del sale di fosfato hanno prodotto 6.
Un gruppo fosfonato è un bioisostere di un gruppo carbossilato con un'acidità maggiore. Le interazioni ioniche tra i residui fosfonato e quelli basici delle proteine bersaglio sono generalmente più forti di quelle dei carbossilati. L'analogo fosfonato 132 è stato sintetizzato da 131b tramite idrogenolisi dell'azide seguita da deprotezione del fosfonato usando TMSBr (Schema 15). In aggiunta, gli analoghi 134 e 135 contenenti un gruppo guanidina, invece del gruppo amino in C-5, sono stati sintetizzati da 131 (Schema 16).
Quindi, l'idrogenolisi dell'azide e la successiva formazione della guanidina e deprotezione hanno portato all'analogo carbossilato 134 da 131a, e l'analogo fosfonato 135 da 131b.
Schema 15.
La sintesi iniziava dal derivato del D-xylosio 122. Dopo protezione selettiva del gruppo idrossi primario con una funzionalità pivaloile, il gruppo idrossi secondario è stato convertito in un α-amino gruppo (123) attraverso ossidazione, formazione dell'ossima, e riduzione.
N-acetilazione, introduzione del gruppo benzilossi in posizione anomerica in condizioni acide, e formazione dell'acetale hanno prodotto 124. La parte fosfato è stata introdotto attraverso attivazione del gruppo idrossi primario come triflato, seguita da sostituzione tramite l'anione fosfonato derivato da 125 o 126, producendo 127a e 127b, rispettivamente. Il gruppo benzile anomerico è stato rimosso idrogeneticamente, ed il nucleo cicloesenico dei composti d'interesse è stato costruito tramite un reazione intramolecolare di Horner–Wadsworth–Emmons, portando al 128.
I rimanenti compiti a questo stadio erano: (1) conversione dell'OH in C-5 in una funzionalità amino, e (2) introduzione del gruppo 3-pentilossi in C-3.
La conversione dell'OH in C-5 in una funzionalità amino è stata ottenuta attraverso inversione di Mitsunobu del gruppo β-idrossi in C-5 con DPPA, diisopropil azodicarbossilato (DIAD), e Ph3P[39]. Dopo idrolisi dell'acetale in condizioni acide, il gruppo idrossi in C-3 è stato invertito attraverso formazione del triflato seguita da attacco SN2 con KNO2. Il gruppo 3-pentile è stato quindi introdotto attraverso sostituzione del 3-pentilossi immidato attraverso l'ossigeno in C-3 in presenza di TfOH. Infine, l'idrogenolisi dell'azide in presenza del catalizzatore di Lindlar e la successiva formazione del sale di fosfato hanno prodotto 6.
Un gruppo fosfonato è un bioisostere di un gruppo carbossilato con un'acidità maggiore. Le interazioni ioniche tra i residui fosfonato e quelli basici delle proteine bersaglio sono generalmente più forti di quelle dei carbossilati. L'analogo fosfonato 132 è stato sintetizzato da 131b tramite idrogenolisi dell'azide seguita da deprotezione del fosfonato usando TMSBr (Schema 15). In aggiunta, gli analoghi 134 e 135 contenenti un gruppo guanidina, invece del gruppo amino in C-5, sono stati sintetizzati da 131 (Schema 16).
Quindi, l'idrogenolisi dell'azide e la successiva formazione della guanidina e deprotezione hanno portato all'analogo carbossilato 134 da 131a, e l'analogo fosfonato 135 da 131b.
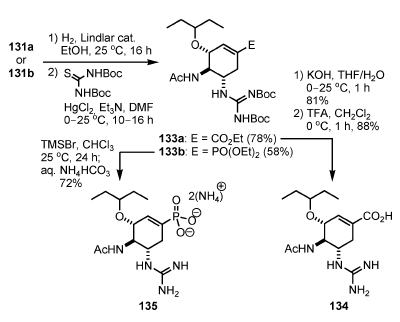 Scheme 16. Sintesi dell'analogo di Fang.
La comparazione delle attività inibitorie di 7, 132, 134, e 135 verso le neuraminidasi bersaglio dei virus dell'influenza di tipo selvaggio (H1N1 e H5N1) e la neuraminidasi dei virus mutanti resistenti agli inibitori (H274Y) sono mostrati nella Tabella 1.
L'analogo 135, contenente gruppi fosfonati e guanidina, ha mostrato in particolare una maggiore potenza rispetto a 7. L'analogo 135 ha anche inibito enzimi mutanti a basse concentrazioni nM.
Sebbene l'efficienza sintetica non fosse necessariamente alta, questo lavoro è degno di nota perché nuovi potenti inibitori della nueraminidasi
vengano identificati durante lo sviluppo di una nuova via sintetica.
Scheme 16. Sintesi dell'analogo di Fang.
La comparazione delle attività inibitorie di 7, 132, 134, e 135 verso le neuraminidasi bersaglio dei virus dell'influenza di tipo selvaggio (H1N1 e H5N1) e la neuraminidasi dei virus mutanti resistenti agli inibitori (H274Y) sono mostrati nella Tabella 1.
L'analogo 135, contenente gruppi fosfonati e guanidina, ha mostrato in particolare una maggiore potenza rispetto a 7. L'analogo 135 ha anche inibito enzimi mutanti a basse concentrazioni nM.
Sebbene l'efficienza sintetica non fosse necessariamente alta, questo lavoro è degno di nota perché nuovi potenti inibitori della nueraminidasi
vengano identificati durante lo sviluppo di una nuova via sintetica.
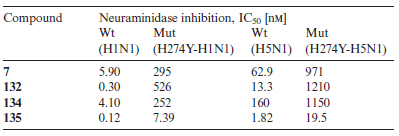 Tabella 1. Attività inibitorie verso le neuroaminidasi di virus influenzali wild-type (Wt) e mutanti (Mut).
Tabella 1. Attività inibitorie verso le neuroaminidasi di virus influenzali wild-type (Wt) e mutanti (Mut).