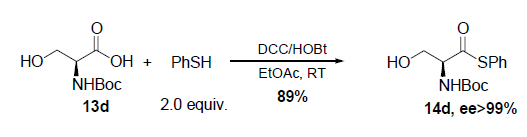quimico
2012-06-09 09:51
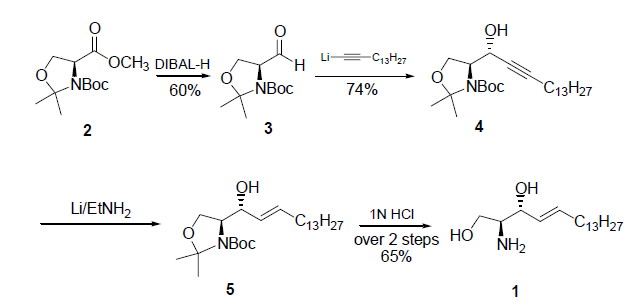
Riprendo qui il discorso fatto altrove sulla sfingosina, ma ovviamente in termini puramente sintetici. Riporto per il momento una delle vie di sintesi.
Ce ne sono molte altre ovviamente.
Vediamo di fare prima una premessa.
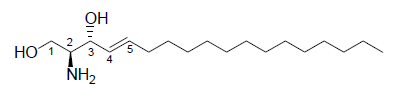 (1)
(1)
Gli sfingolipidi che si ritrovano in natura sono importanti lipidi derivati strutturalmente dalla (-)-D-eritro-sfingosina (1). Gli sfingolipidi sono composti da tre classi di lipidi: ceramidi, sfingomieline e glicosfingolipidi1.
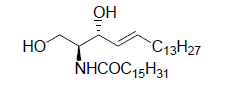 ceramidi
ceramidi
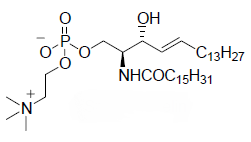 sfingomieline
sfingomieline
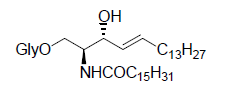 glicosfingolipidi
glicosfingolipidi
Gli sfingolipidi possono essere isolati da membrane cellulari e tessuti neurali. Le loro attività biologiche sono state associate al controllo della crescita, alla maturità, alla sopravvivenza, e alla morte delle cellule. E in modo sorprendente, gli sfingolipidi hanno mostrato una promettente efficacia nella soppressione della crescita delle cellule cancerose2. In aggiunta allo scopo di curare male, le relative N-acilsfingosine (ceramidi) sono già diffusamente usate in campo cosmetico come ingredienti attivi per aumentare la coesione cellulare della pelle3.
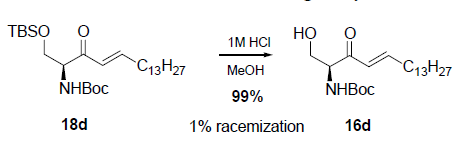
Le attività biologiche versatili degli sfingolipidi rendono quest'ultimi una ricerca preziosa per la chimica di sintesi. Sin dalla risoluzione della struttura della D-eritro-sfingosina fatta da Jenny e Grob4, più di 50 sintesi della sfingosina sono state divulgate5. Di queste, gli approcci più economici usano la L-serina come materiale di partenza6. Il beneficio di questa strategia trae origine dalla presenza di un gruppo idrossile e di un gruppo amino così come dalla presenza di uno stereocentro sulla serina. Comunque, la facilità con cui la serina racemizza sia in condizioni acide sia in condizioni basiche limita la sua applicazione alla sintesi della (-)-D-eritro-sfingosina di elevata enantiopurezza (ee >99%).
Garner et al. hanno provato una sintesi corta ed efficace della sfingosina tramite un'addizione stereocontrollata di un alchinil litio alla ossazolidin aldeide 3, un intermedio chiave, seguita dalla riduzione nelle condizioni di Benkeser usando litio in etilamina ed infine da deprotezione7. Comunque, è stato difficile ottenere l'intermedio chiave 3 con un'enantiopurezza davvero elevata (solo 95-98%)8.
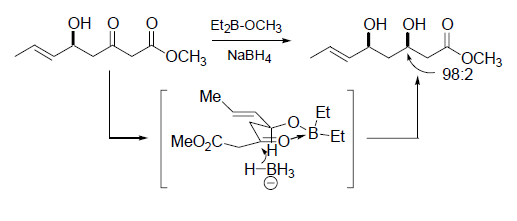
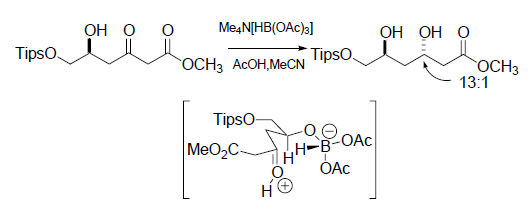
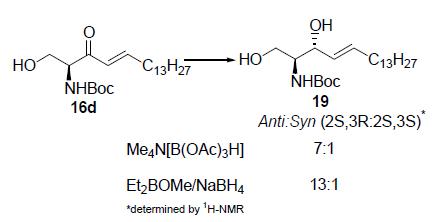
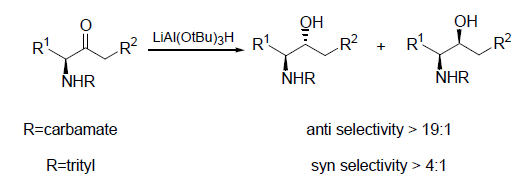
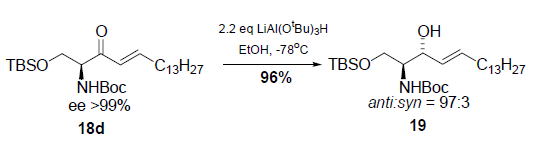
Una strategia più pratica riportata da Koskinen9 traeva vantaggio dalla reazione di Horner-Wadsworth-Emmons per preparare un enone 7 da un chetofosfonato molto enantiopuro derivato dalla L-serina 6. Sebbene i tentativi iniziali comportavano racemizzazione dell'enone 7 prodotto, è stato trovato che K2CO3/MeCN era un ottimo sistema base/solvente per superare il problema della racemizzazione.
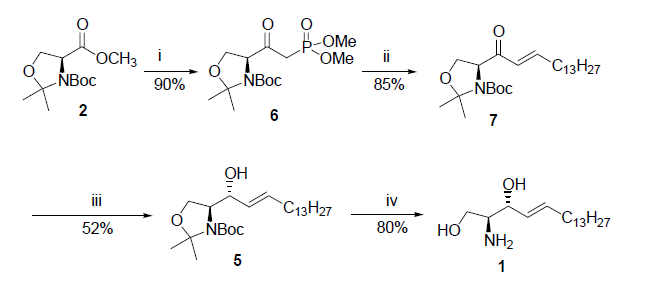
Condizioni: (i) n-BuLi/Dimetilmetilfosfonato/THF (ii) C13H27CHO/K2CO3/MeCN (iii) L-Selectride/THF (iv) 1N HCl
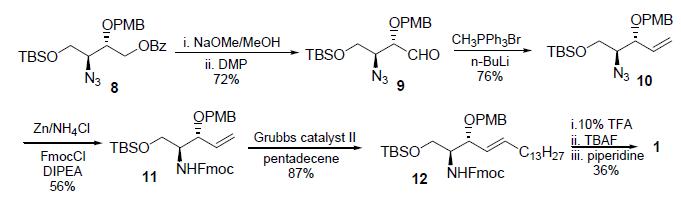
Quale importante metodo di olefinazione, la cross metathesis è stata dimostrato da Somfai10 e Basu11 essere un'efficace strategia per sintetizzare la sfingosina a causa della sua delicatezza ed elevata selettività che ha portato solo all'alchene trans. Comunque, un eccesso di pentadecene è stato incorporato per evitare l'omo-metatesi dei substrati.
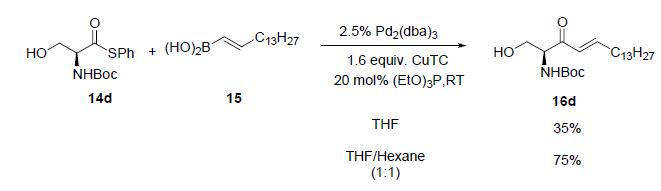
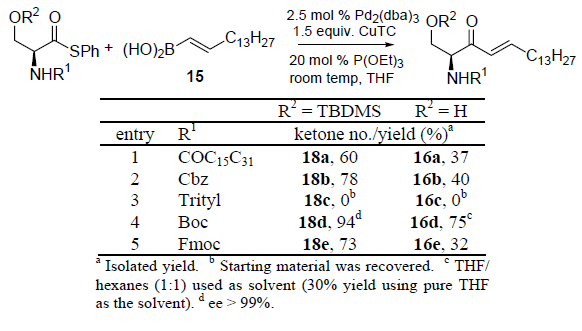
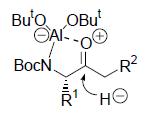
Liebeskind et al. hanno inventato una sintesi estremamente blanda di chetoni tramite il cross coupling di tiolo esteri ed acidi boronici12. Questa nuova reazione utilizza palladio(0) catalitico per attivare gli esteri tiolici tramite un'iniziale addizione ossidativa del legame carbonile-zolfo. Il risultante complesso R1COPdL2SR possiede un forte legame Pd-S che permette solo a forti reagenti nucleofili come gli organolitio, -magnesio o -zinco di effettuare una transmetallazione ed una eliminazione riduttiva che conduce al prodotto, il chetone13. L'introduzione di un carbossilato di Cu(I) nella reazione di Liebeskind-Srogl fornisce una duplice attivazione dei tiol esteri e degli acidi boronici tramite interazioni soft-soft (S-Cu) e hard-hard (O-B), che sono mostrate sotto come stati di transizione proposti per questa reazione. Con questa duplice attivazione tramite un carbossilato di Cu(I), i tiol esteri si accoppiano efficacemente con gli acidi boronici o con gli organostannani producendo chetoni.
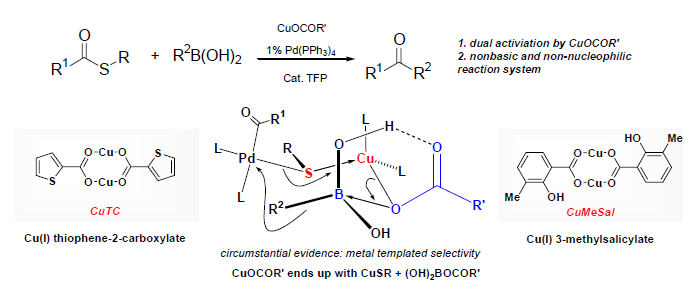
Basandosi sullo stesso principio, il gruppo di Liebeskind ha sviluppato una serie di cross-coupling basati sugli organozolfo per sintetizzare alchini14, nitrili15, eterocicli sostituiti16, ammidine funzionalizzate17 così come pirimidine sostituite tramite un interessante catalizzatore variabile18.
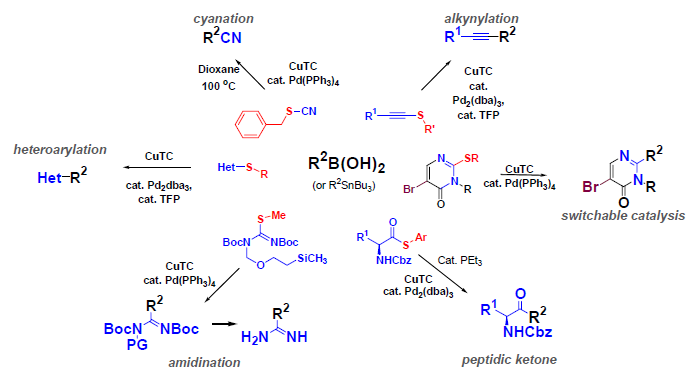
Recentemente, il laboratorio di Liebeskind ha esteso questo principio sviluppando una sintesi di chetoni peptidici con enantipurezza elevate19. Lo sviluppo fortunato di questa metodologia è stato basato sullo screening di diversi ligandi di supporto. L'aggiunta di un trietil fosfito (POEt3) come ligando di supporto è stato in grado di sopprimere la reazione parassita di decarbonilazione. Quale approccio davvero blando, questa reazione è stata condotta a temperatura ambiente in assenza di base. Questa metodologia neutra ed efficace è stata in grado di produrre diversi α-amino chetoni e chetoni peptidici con elevate enantiopurezze senza racemizzazione.
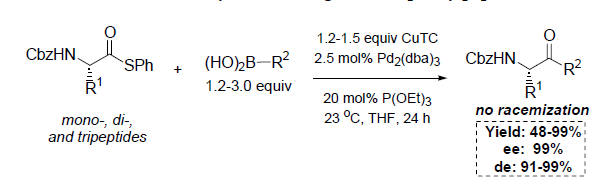
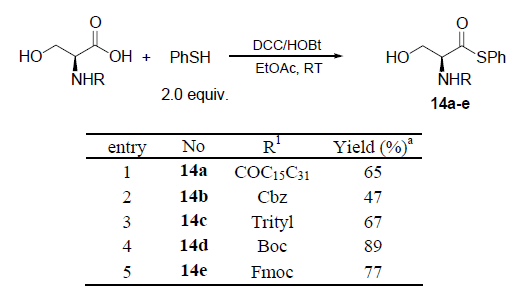
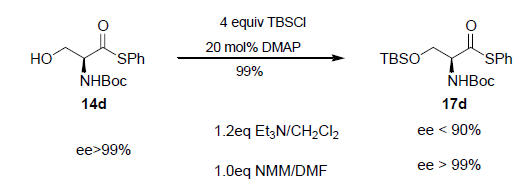
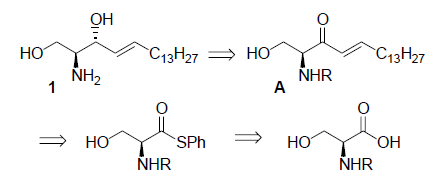
L'analisi retrosintetica della (-)-D-eritro-sfingosina è presentata qui sotto. La chiave per generare la sfingosina ad elevata enantiopurezza dalla L-serina è l'efficace costruzione di un enone A senza racemizzazione che può essere realizzata tramite cross coupling del tiol estere della serina con l'acido trans-1-pentadecenil boronico.
I seguenti utenti ringraziano quimico per questo messaggio: Mario, al-ham-bic