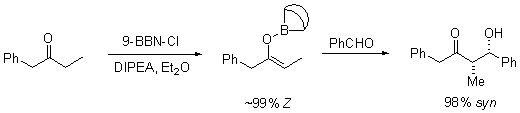
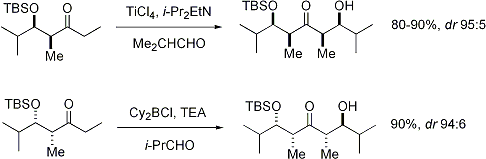
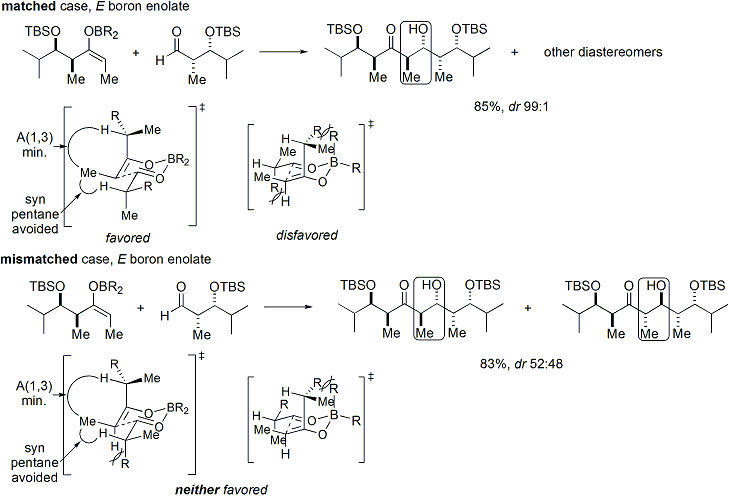
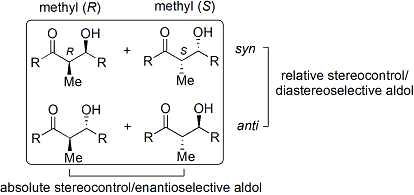
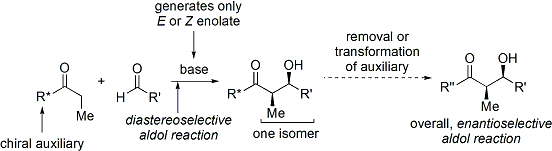
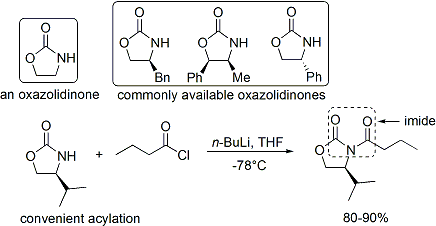
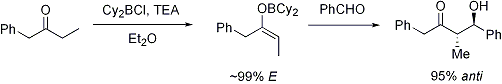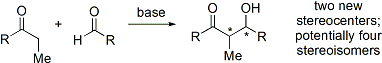Reazioni di specie tipo-enolato
1. Regioselettività nella formazione dello ione enolato e reazione
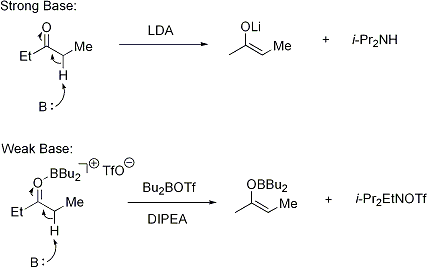
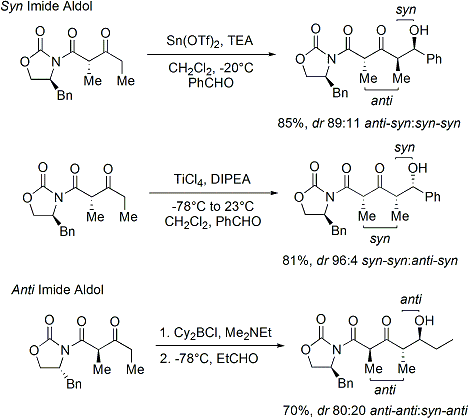
L'importanza degli anioni enolati quali intermedi sintetici è ben stabilita. Malgrado ciò, rimangono dei problemi riguardo la loro selettiva formazione e reazione. Per esempio, le basi enolato di aldeidi sono adatte per subire la reazione aldolica durante la loro formazione, ed i chetoni tipo il 2-eptanone hanno due carboni α, ognuno capace di enolizzazione. La natura ambidentata degli anioni enolato permette anche l'attacco elettrofilo sia all'ossigeno sia al carbonio, ma nella maggior parte delle applicazioni sintetiche è desiderato il legame col carbonio. Infine, gli anioni enolato possono spesso essere formati come stereoisomeri E/Z, ed è stato mostrato che la stereoselettività della reazione, quando vengono creati nuovi centri chirali, dipende dalla configurazione dell'enolato.
Il seguente schema illustra come le condizioni in cui la formazione dell'anione enolato è realizzata possano influenzare la regioselettività della reazione. I due substrati chetonici, il 2-eptanone ed il 2-metilcicloesanone, ognuno possiede carboni α differentemente sostituiti. In ogni caso caso, le miscele di anioni enolato sono generate tramite reazione con una forte base ammide secondaria (LDA è la tipica scelta). Se il chetone è aggiunto ad una soluzione fredda in THF di un eccesso di base, la formazione dell'anione enolato è veloce ed irreversibile (procedura a). D'altra parte, se un legger eccesso del chetone è lasciato rimanere in soluzione, un equilibrio che coinvolge il chetone e le diverse specie enolato viene stabilita (procedura b). All'equilibrio il più stabile anione enolato predominerà. Gli esempi riportati nello schema riportano anche i risultati da una preparazione all'equilibrio in cui il metallo litio nell'LDA è sostituito dal potassio (procedura c).
Formazione regioselettiva degli anioni enolato
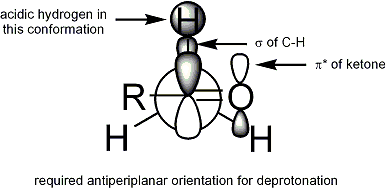
Diversi importanti principi sono dimostrati qui. Primo, se la specie enolato ha un sostanziale carattere di doppio legame, il doppio legame maggiormente sostituito dovrebbe predominare all'equilibrio, come previsto dalle stabilità degli alcheni sostituiti. Dato che i legami litio-ossigeno sono più covalenti (hanno meno carattere ionico) rispetto ai legami potassio-ossigeno, l'enolato di litio si avvicina maggiormente ad un alchene rispetto all'enolato di potassio. Secondo, il maggiore carattere ionico dell'enolato di potassio pone una carica negativa aumentata sul carbonio α, una condizione che è sfavorita dalla sostituzione del gruppo alchile. Inoltre, l'ordine di stabilità dei carbanioni sostituiti è inversa a quella dei carbocationi grazie al carattere elettron donatore dei gruppi alchilici rispetto all'idrogeno. Infine, la velocità della rimozione del protone da un carbonio α è diminuita dalla sostituzione alchilica, probabilmente a riflettere una combinazione di ingombro sterico (basi ingombrate) e stabilità carbanionica diminuita. In entrambi gli esempi mostrati sopra, le condizioni usate nella procedura (a) sono tipiche della formazione dell'enolato cineticamente favorita, mentre quelle usate nella procedura (b) favoriscono la formazione termodinamica dell'enolato. Le acidità relative fornite dai valori di pKa sono derivate da misure in condizioni all'equilibrio, e riflettono quindi l'acidità termodinamica. Le determinazioni di acidità cinetica richiedono esperimenti di scambio isotopico competitivo.
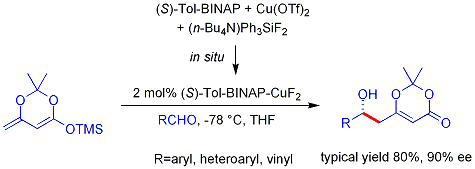
Questi principi influenzano il decorso delle reazioni di alchilazione di enolati, come mostrato nel seguente schema. Nel primo caso, il 2-metilcicloesanone viene convertito in una miscela termodinamica di enolati, che viene quindi fatta reagire con metile ioduro. Il prodotto maggioritario è il previsto 2,2-dimetilcicloesanone (dal più stabile anione enolato), ma questo viene accompagnato da prodotti di- e trimetilati assieme a circa il 20% di materiale di partenza non reagito. La complessità della miscela di prodotti è dovuta al trasferimento acido-base del protone tra i prodotti alchilati e l'anione enolato non reagito. In altre parole, non appena una piccola quantità (diciamo il 5%) di dimetilcicloesanone si è formata, essa trova sé stessa in soluzione con una concentrazione relativamente elevata di una base forte (il rimanente anione enolato) che può rimuovere un'altro protone α, dando un nuovo anione enolato che è successivamente metilato. Se l'enolato di litio cineticamente favorito è usato invece degli enolati di potassio all'equilibrio, il 2,6-dimetilcicloesanone è il prodotto principale.
La seconda reazione è un'alchilazione intramolecolare che può avvenire in due modi diversi. Se l'enolato cineticamente favorito (rimozione del protone del metile) è formato a bassa temperatura, esso reagisce per riscaldamento formando un anello a sette membri. Alternativamente, la base più debole, potassio t-butossido (in t-butanolo come solvente), genera una miscela all'equilibrio di enolati che eventualmente reagiscono tramite alchilazione intramolecolare. L'α'-enolato termodinamicamente favorito predomina, e l'alchilazione risultante genera un anello a cinque membri.
Esempi di alchilazione selettiva di enolati
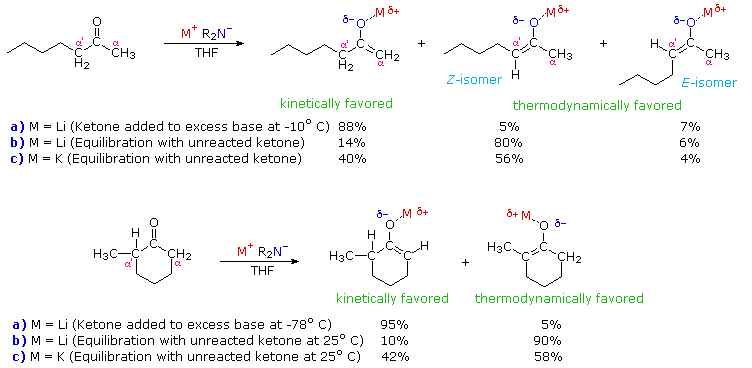
Un altro aspetto dell'alchilazione di un anione enolato, non ancora affrontato, è la possibilità di legame elettrofilo all'ossigeno. Un esempio di tale comportamento è mostrato sotto.
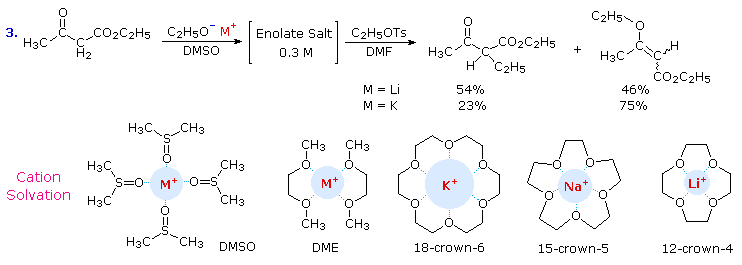
A causa della sostanziale carica negativa sull'ossigeno degli anioni ambidentati, ci si potrebbe aspettare che l'O-alchilazione sia una regola piuttosto che un'eccezione. Questo, infatti, è vero quando sali di enolati totalmente o ampiamente ionizzati vengono fatti reagire con forti elettrofili. La ionizzazione degli enolati è facilitata da solventi con alte costanti dielettriche, come il DMSO e al DMF, specialmente per i sali con i cationi potassio e cesio. Come mostrato nella parte inferiore del secondo schema, gli ossigeni negativamente caricati del DMSO si raggruppano attorno ad un catione, fornendo sostanziale stabilizzazione per solvatazione. Una tale solvatazione per l'anione enolato non esiste, lasciandolo aperto alla reazione con un elettrofilo. Gli enolati di litio hanno un carattere significativamente covalente nel legame metallo-ossigeno, e questo ritarda l'attacco elettrofilo all'ossigeno.
Solventi eterei come il THF ed il DME (dimetossietano o glyme) sono comunemente ustai per le alchilazioni perché sono inerti alla basi forti e dissolvono i sali di enolato più efficacemente degli idrocarburi. L'etere difunzionale DME è specialmente efficace nel solvatare i cationi; e questo fatto ha portato alla preparazione di polieteri ciclici, noti come eteri corona, che sono agenti solvatanti estremamente potenti. Gli eteri corona possono essere aggiunti a soluzioni del sale di enolato per aumentarne la loro ionizzazione. Inoltre, la dimensione dell'etere corona può essere fatto su misura per adattarsi al catione che è stato usando, fornendo addizionale controllo sul decorso delle reazioni di enolati.
2. Preparazione e reazioni dei silil enol eteri
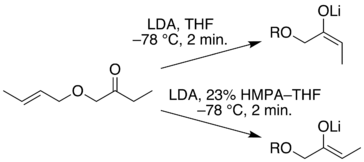
Un modo di preparare intermedi anioni enolati selettivi è di prima intrappolare ed isolare essi come silil enol eteri. Questi composti relativamente stabili possono quindi essere usati per generare anioni enolato isomericamente puri, o in alcuni casi come nucleofili enolici. Nello schema seguente, la prima reazione illustra la formazione di una miscela di silil enol eteri in condizioni all'equilibrio. Se un'elevata proporzione dell'isomero minoritario è desiderata, l'enolato di litio cineticamente favorito può essere preparato e quenchato con trimetilsilil cloruro. In entrambi i casi la miscela del silil etere può essere separata per distillazione. Una volta che l'isomero silil etere puro è in mano, esso può essere usato per generare il corrispondente enolato di litio nella maniera mostrata. Le reazioni di alchilazione di questi enolati produce quindi prodotti puri regioisomerici.
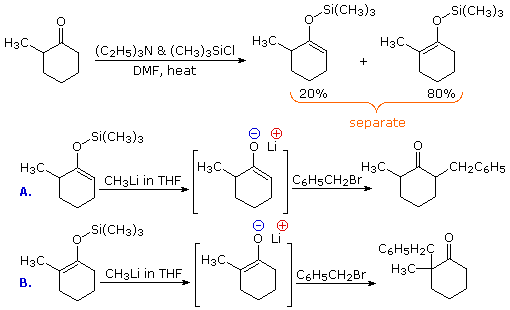
Sono sono riportati altri due esempi dell'uso diretto dei silil enol eteri.
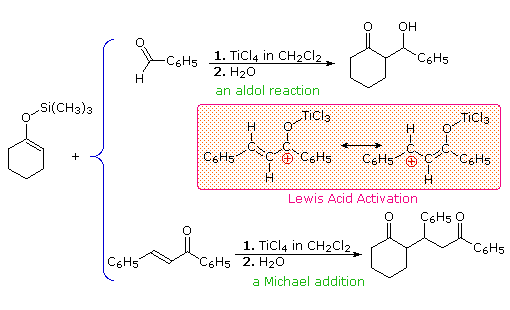
Dato che i silil eteri non sono reattivi come gli anioni enolati, gli elettrofili con cui essi si combiano deve essere resi più reattivi. Quando sono usati carbonili elettrofili, questi può essere realizzato tramite catalizzatori acidi di Lewis, come mostrato.
3. Enammine come surrogati dell'anione enolato
La formazione di enammine tramite reazione di amine secondarie con aldeidi o chetoni verrà descritta in modo più approfondito a breve. Il doppio legame dell'enammina trasmette il carattere nucleofilo dell'azoto al carbonio α, in un modo vinilogo. A causa della risultante nucleofilicità ambidentata delle enammine, le reazioni con gli elettrofili possono avvenire sia all'azoto che al carbonio. Le enammina derivata dalle aldeidi sono di solito alchilate all'azoto, un decorso non desiderabile per la maggior parte delle applicazioni sintetiche. I chetoni danno significativa
C-alchilazione, il decorso termodinamicamente favorito, come dimostrato da G. Stork (Columbia). Lo ione imminio creato tramite
C-alchilazione non puà reagire ulteriormente, ed è facilmente idrolizzato a chetone alchilati. Questo è particolarmente utile se i prodotti di dialchilazione devono essere evitati. Perciò, nel primo esempio, la metilazione diretta dell'anione enolato da questo chetone dà quantità significative del prodotto dimetile, a causa dello scambio di protone dell'enolato. Come mostrato, la via tramite enammina dà solo il prodotto mono-metilato.
Il secondo esempio dimostra che le enammine possono essere acilate bene così come alchilate. Infatti, la natura reversibile dell'acilazione rimuove il problema della competitiva
N-acilazione. Questo caso illustra anche la tendenza generale a formare l'enammina meno sostituita quando due diversi siti α sono presenti case also illustrates the general tendency to form the least substitute. La coniugazione della coppia di elettroni non legante con gli elettroni π del doppio legame forza i sostituenti alchilici sull'azoto a giacere nello stesso piano come il doppio legame. Come risultato la sostituzione del doppio legame porta ad un aumento dell'ingombro sterico con i sostituenti all'azoto. L'amina ciclica secondaria a cinque membri pirrolidina è diffusamente usata per le reazioni dell'enammine, in parte perché quest ingombro sterico è minimizzato.
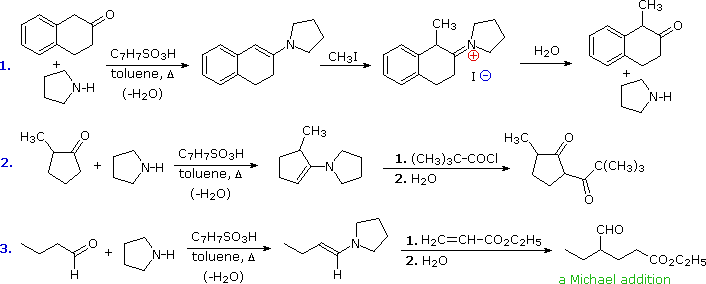
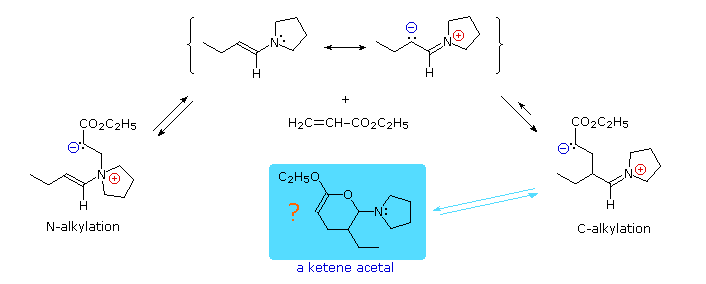
Come notato, l'
N-alchilazione delle enammine è comune per le aldeidi ed alcuni chetoni. Le reazioni di addizione di Michael evitano questo problema grazie alla loro reversibilità. Il terzo esempio mostra una tale reazione, e sotto viene mostrato anche un possibile meccanismo. L'intermedio derivato dalla
C-alchilazione è termodinamicamente più stabile della specie derivata dalla
N-alchilazione, così esso predomina all'equilibrio. Entrambe le cariche in questo intermedio sono stabilizzate tramite delocalizzazione, e l'idrolisi rapidamente lo converte nel prodotto aldeide-estere. Una interessante alternativa è la chiusura d'anello a composto enol etere neutro (mostrato nel riquadro blu ombreggiato) che dovrebbe anche essere idrolizzato allo stesso prodotto.
 4. Anioni di immine ed idrazoni
4. Anioni di immine ed idrazoni
Un'altra via di aggirare alcuni degli aspetti indesiderati della chimica degli anioni enolato è sostituire l'ossigeno di una aldeide o chetone con un amino gruppo primario, in altre parole, convertire la funzione carbonilica in una immina. I derivati imminici sono relativamente facili da preparare, a partire da un'aldeide o chetone e da un'amina primaria o da un'idrazina. La funzione C=N risultante non attiva i gruppi α-C-H così efficacemente come una funzione carbonilica, ma basi davvero forti come l'LDA, gli alchillitio ed i reagenti di Grignard convertiranno le immine nelle loro basi coniugate enammidi in modo quantitativo. Questa reazione generale è mostrata nel riquadro verde ombreggiato sotto.
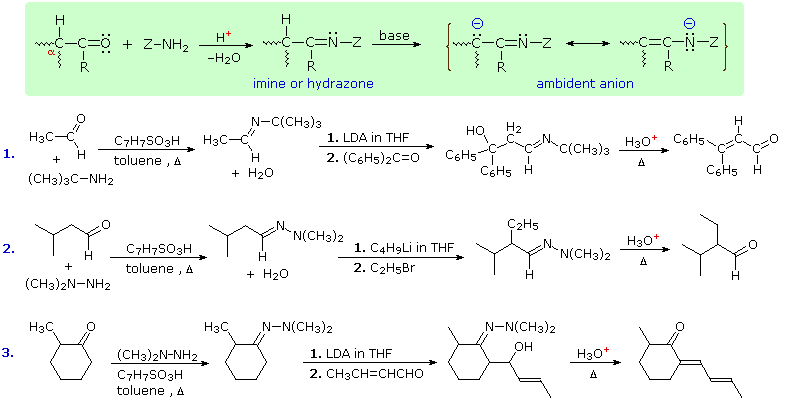
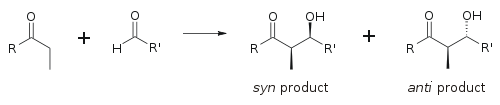
Tre schemi dell'uso delle basi enammidiche nella sintesi sono mostrati sotto. I primi due esempi usano derivati aldeidici, e se si tentasse di fare queste reazioni con l'anione enolato dell'aldeide solamente, ne risulterebbe una dimerizzazione dell'aldolo. La funzione C=N delle immine è un povero accettore di nucleofili, così non assume un tale ruolo nella reazioni tipo aldoliche. La terza reazione è una condensazione aldolica in cui un chetone serve da donatore. Se dei sali di rame(I) sono introdotti prima che l'aldeide insatura è aggiunta alla soluzione dell'enammide, avviene una addizione coniugata in preferenza all'addizione 1,2-aldolica.



 o usando un acido di Lewis e un base debole ("condizioni soft"
o usando un acido di Lewis e un base debole ("condizioni soft"