Myttex Forum ha chiuso definitivamente. Non è più possibile inviare messaggi, ma il contenuto è ancora consultabile in questo archivio.
ohilà
2015-04-21 14:08
Com'è noto gli alcoli terziari possono essere clorurati facilmente anche dal semplice acido cloridrico. Visto che avevo un po' di terz-butanolo, ho provato questa semplice sintesi.
REAGENTI
- Alcol t-butilico
- Acido cloridrico 36%
- Bicarbonato di sodio
- Solfato di magnesio anidro
Il t-butanolo ha un p.f. di soli 25°C, quindi a temperatura ambiente è un solido untuoso mezzo sciolto. Il modo migliore per maneggiarlo è portarlo a fusione completa scaldandolo un pochino con un phon.
A contatto con la vetreria fredda cristallizza rapidamente.
reag.jpgt-buoh crist.jpg
PROCEDURA
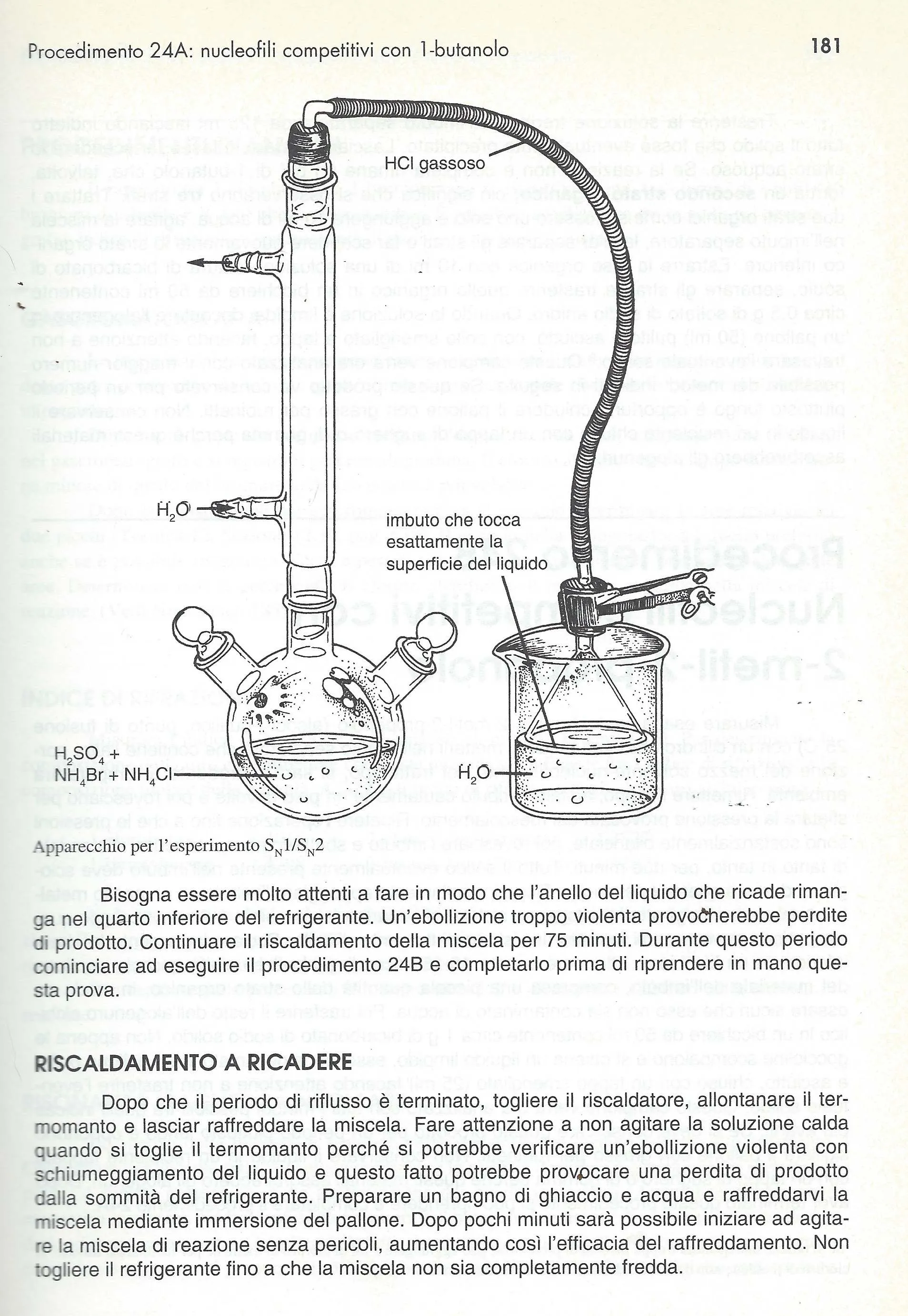
Porre su agitatore una miscela di 35 grammi di alcol e 120 ml di HCl concentrato. Agitare per venti minuti e poi versare la miscela in un imbuto separatore. Si conserva la fase superiore.
agitazione.jpgimbuto.jpg
Si lava con 30 ml di NaHCO3 5% e poi con 20 ml d'acqua. Distillare raccogliendo tra 49 e 51°C usando una piastra (no fiamma!) e avendo cura che l'acqua del liebig sia sempre ben fredda.
Rimane qualche gocciolina di "coda", evidentemente un po' d'acqua o alcol residui.
distillazione TBUCL.jpg
Seccare su solfato di magnesio anidro. Si ottengono 35 ml circa di cloruro.
fine.jpgformula.png
I seguenti utenti ringraziano ohilà per questo messaggio: fosgene, quimico, luigi_67, zodd01, Hamiltoniano, ale93
quimico
2015-04-21 15:51
Bravo. Interessante sintesi, ottima per farsi la mano.
Dovresti provare a farne altri, magari più complessi, potendo ovvio.
Bravo. Interessante sintesi, ottima per farsi la mano.
Dovresti provare a farne altri, magari più complessi, potendo ovvio.
Hai in mente di farci qualcosa?
Il t-butanolo in realtà l'avevo preso per fare il potassio col metodo di Kirmer.
Ma intanto ho deciso di usarne un po' per farci il cloruro, che, però, per adesso, non mi serve a niente...
Eh, per farne altri servirebbe il tionilcloruro... O forse si può provare con ZnCl2/HCl, ma non sembra troppo facile... Boh, vedremo...
fosgene
2015-08-04 14:04
Sintesi effettuata a scuola nel seguente modo:
In un imbuto separatore da 250mL si pongono 20mL di tert-butanolo e 50mL di HCl 37%. Quindi si agita delicatamente l' imbuto con la miscela per 20 minuti sfiatando spesso per evitare soprapressioni.
1.JPG
Al termine della reazione si saranno formati due strati ben distinti:
uno superiore torbido composto da cloruro alchilico + acqua e uno inferiore formato da acqua e acido non reagito:
2.JPG
Si scarta lo strato acido inferiore e si lava il cloruro alchilico grezzo con una soluzione al 5% m/V di NaHCO3. Durante questo passaggio si sviluppa molta CO2 evitare di tappare ermeticamente l' imbuto per non creare sovrappressioni.
Una volta cessata l' effervescenza si scarta la fase acquosa neutralizzata e si lava la fase rimanente con acqua distillata. Al termine di questo passaggio il nostro prodotto si essicca in una beutina con solfato di sodio anidro fino a totale limpidezza.
Si mette il nostro prodotto in un palloncino da 50mL e si collega a un apparato per la distillazione frazionata. Tra il palloncino e il condensatore va inserita una colonna di Vigreux per la separazione dell fasi volatili da quelle meno volatili. Il riscaldamento si effettua con un termomanto.
Si raccoglie solo la frazione che distilla tra 49 e 51°C . Un' eventuale testa e una coda che bollono prima o dopo questo intervallo vanno scartate.
Il palloncino di raccolta va posto in bagno di ghiaccio vista la volatilità del cloruro di ter-butile.
Avevo pure una foto di un grafico IR di un campione abbastanza puro ma per il momento non la trovo, non vorrei averla cancellata per sbaglio
I seguenti utenti ringraziano fosgene per questo messaggio: quimico, luigi_67, ohilà, zodd01
fosgene
2015-08-05 18:16
Ho alcune domande:
Secondo voi, se volessi effettuare la sintesi di questo composto in home-lab, sarebbe migliore la procedura di Ohilà o quella effettuata da me a scuola?
Da quanto mi hanno spiegato l' uso dell' imbuto separatore ha diversi vantaggi:
Essendo provvisto di tappo si evitano perdite di alogenuro per evaporazione e eventuali esalazioni nell' ambiente di lavoro di vapori acidi (HCl).
Vista la sua forma si può scuotere facilmente per ottenere una miscelazione piú intima possibile tra i due reagenti.
A reazione terminata si possono separare subito i due strati liquidi senza effettuare alcun travaso evitando cosí perdite del prodotto per volatilizzazione.
In teoria un beker o una beuta non sarebbero molto adatti a questo scopo.
La colonna di Vigreux é essenziale?
Ci hanno spiegato che oltre alla reazione di SN (che é quella principale) c'é anche una reazione di eliminazione che porta alla formazione di sottoprodotti. In effetti noi abbiamo ottenuto una testa che bolliva intorno ai 20-21°C e una coda altobollente. E ricordo che una delle due aveva un odore oleifinico o comunque caratteristico tipo il gas di bombola.
Non può essere che il prodotto di Ohilà sia contaminato?
marco the chemistry
2015-08-05 18:48
Il modo migliore è porre l'HCl e il t-butanolo in una palloncino, collegare una testa di distillazione e un condensatore e cominciare a scaldare a bagno maria, lentamente distilla l'alogenuro alchilico. In questo modo si ottiene un prodotto puro e nessuna E1.
Dopo questa procedura il liquido viene lavato con acqua e NaHCO3, le rese sono quasi quantitative
Mi spieghi come fai ad affermare che il prodotto di ohilà sia inquinato nonostante lo abbia distillato? usiamo la testa....
I seguenti utenti ringraziano marco the chemistry per questo messaggio: Hamiltoniano
quimico
2015-08-05 18:56
Se una delle due aveva un odore olefinico (?) è perché tramite E1 si ottiene isobutene, un gas. Poi mi spieghi cosa intendi
Concordo con marco. Meglio fare come dice. Si ottiene sicuramente un prodotto puro e senza tanto sbattimento. Tanto devi comunque distillare
Infatti. Il prodotto di ohilà a me pare buono. Non capisco come puoi dire sia inquinato
Mi spieghi come fai ad affermare che il prodotto di ohilà sia inquinato nonostante lo abbia distillato? usiamo la testa....
Infatti l' ho solo supposto, diciamo una presupposizione scritta male
Visto che non aveva usato una Vigreux
marco the chemistry
2015-08-05 19:30
la vigreux è una cosa inutile in questo caso, inutilissima! Un sottoprodotto è un gas, i reagenti bollono molto più alti del prodotto di interesse....
Guglie95
2015-08-06 11:41
Come mai Marco usando la tua procedura non si ottiene E1? Perchè si riscalda lentamente senza raggiungere temperature favorevoli all'eliminazione? Oppure per il particolare procedimento e strumentazioni?
marco the chemistry
2015-08-06 12:15
Perchè ottenere un'eliminazione in queste condizioni è pura fantasia! La reazione di Sn1 è molto più veloce e favorita in condizioni acide, quel poco alchene che eventualmente si forma subisce addizione di HCl..
Per avere facilmente E1 bisogna lavorare in ambiente basico e con l'alogenuro alchilico. Es: t-BuCl + NaOH da credo quasi solo E1 perchè si formano molecole molto stabili come NaCl e H2O, oltre all'ovvio fatto che l'isobutilene prodotto se ne va.
Spero di essere stato chiaro.
I seguenti utenti ringraziano marco the chemistry per questo messaggio: fosgene, luigi_67
quimico
2015-08-06 12:50
Concordo. Le eliminazioni di norma sono favorite quando o si formano prodotti che si possono allontanare dalla reazione come i gas o quando si formano molecole stabili.
Alcune eliminazioni poi sono favorite da basi forti, non nucleofile, ingombrate, basti pensare alla DBU.
Qui l'SN1 è certamente più favorita: si forma un catione terziario stabile, la cinetica della reazione è veloce, il prodotto è stabile... Il sottoprodotto, l'isobutene, formatosi è davvero poco. Mi aspetterei di trovarne davvero tanto ad esempio nel caso dello sblocco del BOC, visto che è anche il fattore che favorisce l'esito quasi quantitativo della deprotezione a dare un prodotto pulito.
I seguenti utenti ringraziano quimico per questo messaggio: fosgene, luigi_67
Guglie95
2015-08-06 13:58
Grazie per le delucidazioni =)
zodd01
2015-10-09 11:25
Pensi possa utilizzare la medesima procedura per il n-butanolo ? Su orgsyn ho trovato solo la ricetta con cloruro di zinco e non lo possiedo.
Perchè ottenere un'eliminazione in queste condizioni è pura fantasia! La reazione di Sn1 è molto più veloce e favorita in condizioni acide, quel poco alchene che eventualmente si forma subisce addizione di HCl..
Per avere facilmente E1 bisogna lavorare in ambiente basico e con l'alogenuro alchilico. Es: t-BuCl + NaOH da credo quasi solo E1 perchè si formano molecole molto stabili come NaCl e H2O, oltre all'ovvio fatto che l'isobutilene prodotto se ne va.
Spero di essere stato chiaro.
Marco, ma l'acidità serve solo a far sputare fuori il gruppo uscente. Una volta che il carbocatione s'è formato si procede velocemente sia con SN1 che con E1, anche se poi il secondo prodotto si volatilizza (quindi come fa a subire addizione!?). SN1 sarà avvantaggiata, ma E1 è comunque presente.
Poi, l'esempio t-BuCl + NaOH. Qui, al contrario, non è che procede più per E1, piuttosto che per SN1 a causa dell'acqua?
I seguenti utenti ringraziano LuiCap per questo messaggio: Guglie95, fosgene, ClaudioG., quimico, zodd01, luigi_67
quimico
2015-10-09 21:36
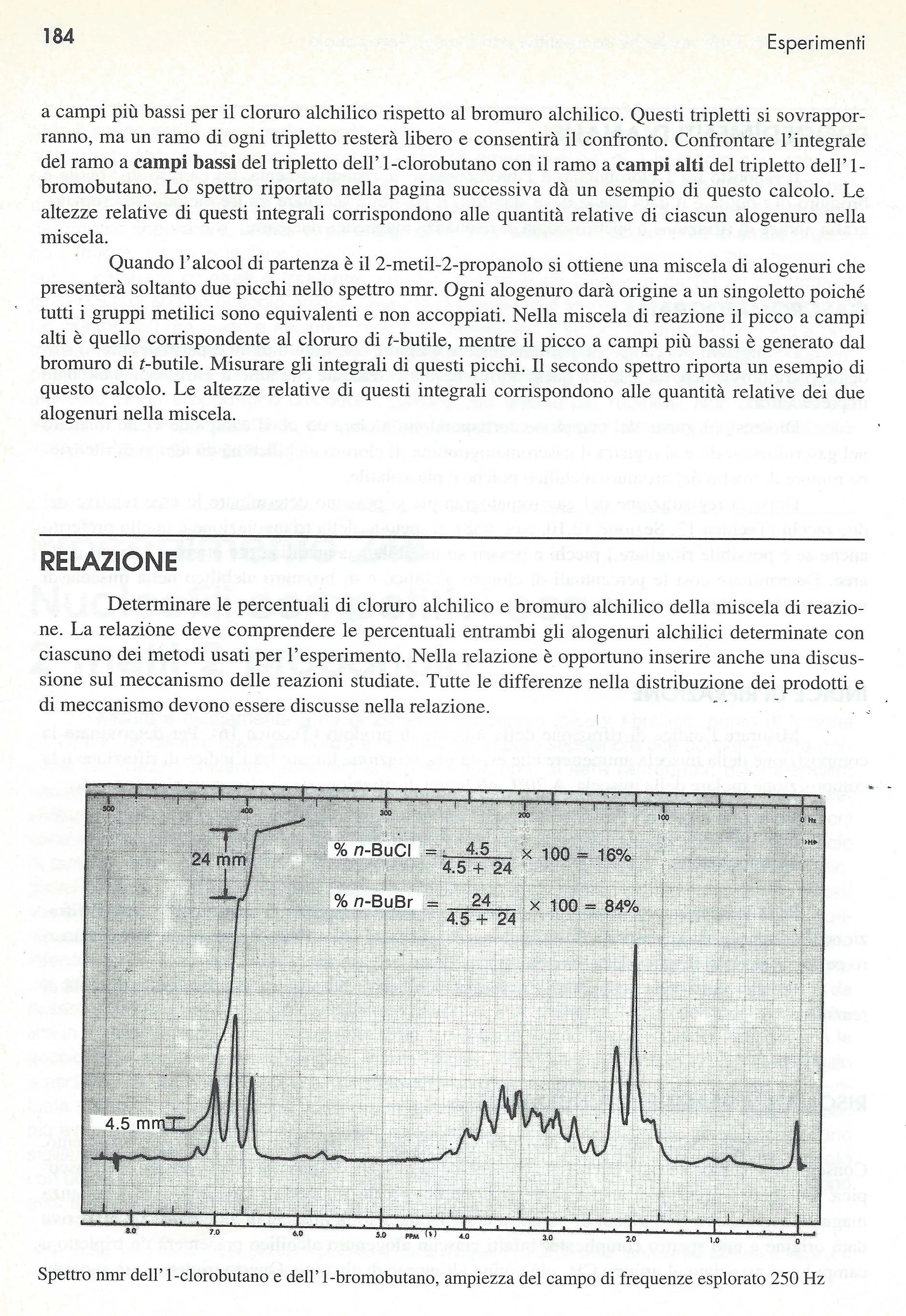
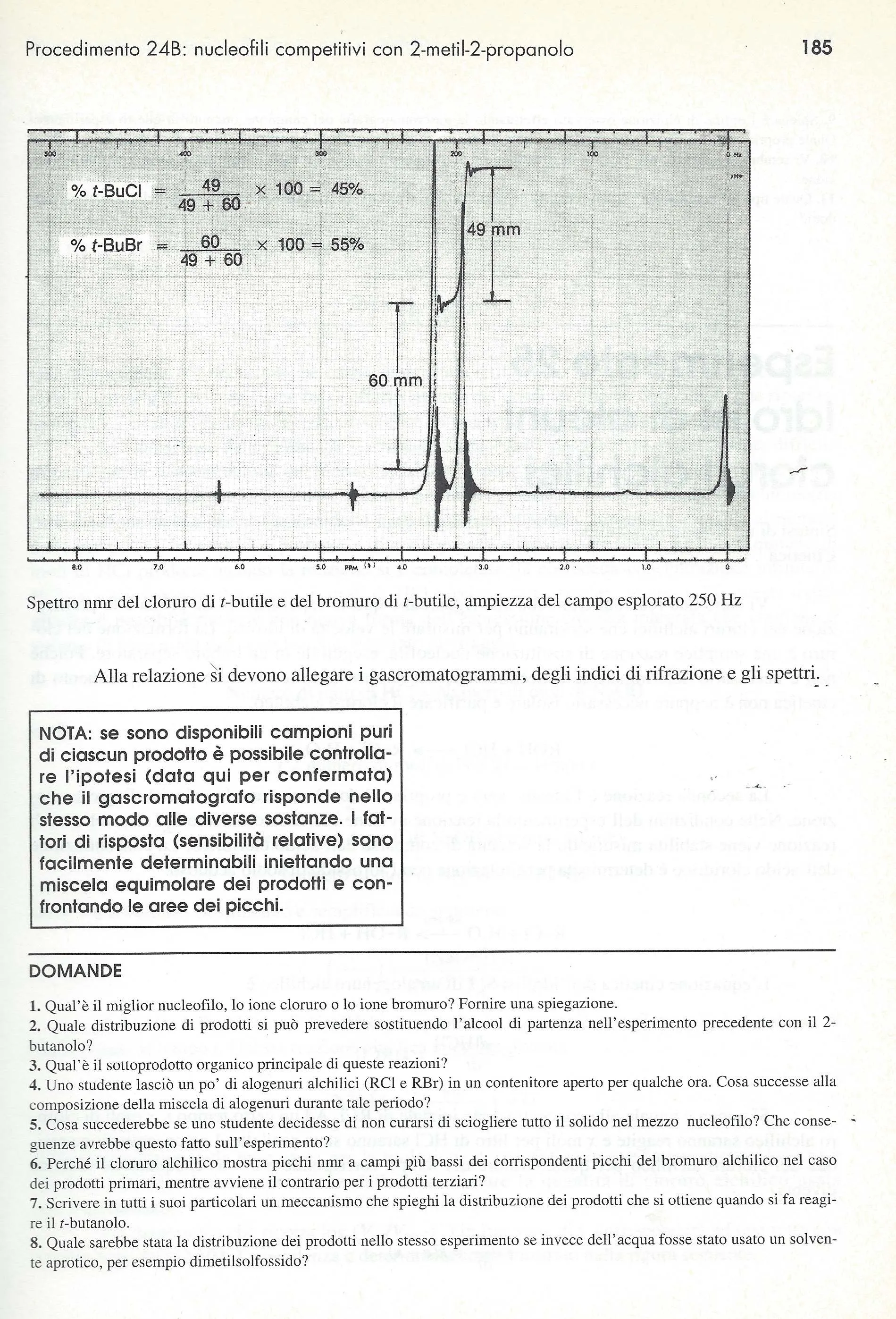
Spettro NMR di altri tempi. Su carta millimetrata a 250 MHz
Grazie Luisa. Molto gentile.
LuiCap
2015-10-09 22:10
Mai fatto uno spettro NMR in vita mia, al massimo arrivo allo spettro IR
Sintesi a volontà, non a livello universitario però, dal 1980 fino al giugno scorso.
In quegli anni, quando le normative in materia di sicurezza erano molto meno restrittive delle attuali, al quarto anno si faceva il Magneson II partendo dal benzene; pochi studenti riuscivano ad arrivare alla fine solo con le quantità dei prodotti delle sintesi intermedie. Non so se fossero più le brutte parole che volavano o le robacce tossiche che si respiravano
zodd01
2015-10-10 06:56
Il magneson dal benzene ? Mamma mia ce ne sarà di lavoro.
Comunque è un piacere avere qui sul forum un insegnante come lei, una persona che ha lavorato per davvero.
Non come alcuni che avevo per professori alla triennale, rinchiusi in teorie complicatissime ( ma inutili ) magari di meccanica quantistica o che usavano l'ultimo modello di supermega diffrattometro a raggi X munito di spara-ioni accelerati e plasma e poi capace non sapevano come fare un bagno acido per lavare la vetreria.
I seguenti utenti ringraziano zodd01 per questo messaggio: LuiCap, quimico















 Visto che non aveva usato una Vigreux
Visto che non aveva usato una Vigreux