quimico
2011-11-26 13:50
Gli isomeri sono composti differenti che hanno la stessa formula molecolare. Quando il gruppo di atomi che costituiscono le molecole di differenti isomeri sono legati insieme in modi fondamentalmente diversi, ci riferiamo a tali composti con il nome di isomeri costituzionali. Per esempio, nel caso degli idrocarburi C4H8, la maggior parte degli isomeri sono costituzionali. Le strutture stenografiche di quattro di questi isomeri sono mostrate sotto con i loro nomi IUPAC.
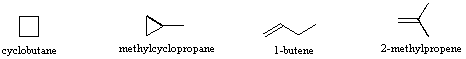 Notare che i dodici atomi che costituiscono questi isomeri sono connessi o legati in modi molto differenti. Così come è vero per tutti gli isomeri costituzionali, ogni composto differente ha un differente nome IUPAC. Inoltre, la formula molecolare fornisce informazioni circa qualcuna delle proprietà strutturali che può essere presente negli isomeri. Dal momento che la formula C4H8 ha due idrogeni in meno dell'alcano butano a quattro atomi d'idrogeno (C4H10), tutti gli isomeri che hanno questa composizione possono incorporare sia un anello che un doppio legame. Un quinto isomero possibile di formula C4H8 è CH3CH=CHCH3. Questo dovrebbe essere denominato 2-butene in accordo con le regole IUPAC; tuttavia, un'ispezione ravvicinata di questa molecola indica che ha due strutture possibili. Questi isomeri possono essere isolati come composti distinti, aventi caratteristiche e proprietà diverse. Essi sono qui mostrati con le designazioni cis e trans.
Notare che i dodici atomi che costituiscono questi isomeri sono connessi o legati in modi molto differenti. Così come è vero per tutti gli isomeri costituzionali, ogni composto differente ha un differente nome IUPAC. Inoltre, la formula molecolare fornisce informazioni circa qualcuna delle proprietà strutturali che può essere presente negli isomeri. Dal momento che la formula C4H8 ha due idrogeni in meno dell'alcano butano a quattro atomi d'idrogeno (C4H10), tutti gli isomeri che hanno questa composizione possono incorporare sia un anello che un doppio legame. Un quinto isomero possibile di formula C4H8 è CH3CH=CHCH3. Questo dovrebbe essere denominato 2-butene in accordo con le regole IUPAC; tuttavia, un'ispezione ravvicinata di questa molecola indica che ha due strutture possibili. Questi isomeri possono essere isolati come composti distinti, aventi caratteristiche e proprietà diverse. Essi sono qui mostrati con le designazioni cis e trans.
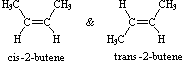
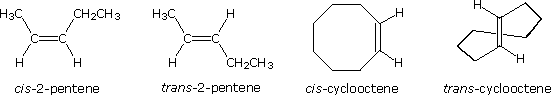
La tipologia di legame degli atomi i questi due isomeri è essenzialmente equivalente, con la sola differenza dovuta alla relativa orientazione o configurazione dei due gruppi metile (e degli atomi d'idrogeno associati) nei confronti del doppio legame. Nell'isomero cis i gruppi metile sono sullo stesso lato, mentre sono sui lati opposti nell'isomero trans. Gli isomeri che differiscono solo nell'orientazione spaziale dei loro atomi componenti sono chiamati stereoisomeri. Gli stereoisomeri richiedono sempre che un prefisso di nomenclatura addizionale sia aggiunto al nome IUPAC allo scopo di indicarne l'orientazione spaziale, per esempio cis (dal latino, che significa: sullo stesso lato) e trans (dal latino, che significa: da una parte all'altra), nel caso del 2-butene. Il doppio legame carbonio-carbonio è formato da due atomi di carbonio sp2 ibridizzati, e consiste di due orbitali molecolari occupati, un orbitale sigma ed un orbitale p-greco. La rotazione dei due gruppi all'estremità di un doppio legame relativamente a tutti gli altri distrugge la sovrapposizione dell'orbitale p creata dall'orbitale o dal legame p-greco. Poiché il legame p-greco possiede una energia di legame pari approssimativamente a 60 kcal/mole, questa resistenza alla rotazione stabilizza la configurazione planare di questo gruppo funzionale. Come risultato si ha che certi alcheni bi-sostituiti possono esistere come una coppia di stereoisomeri configurazionali, spesso designati con cis e trans. Il requisito essenziale per avere questo stereoisomerismo è che ogni carbonio del doppio legame debba avere due differenti gruppi sostituenti (uno dei quali può essere l'idrogeno). Alcuni esempi di questo stereoisomerismo configurazionale (a volte chiamato isomerismo geometrico) sono mostrati appresso. Notare che i cicloalcheni con meno di otto atomi di carbonio non esistono in una configurazione stabile trans, a causa delle tensioni sull'anello. Una simile restrizione riguarda anche i cicloalchini con meno di dieci atomi di carbonio. Dal momento che gli alchini sono lineari, non esiste stereoisomerismo accociato con il triplo legame carbonio-carbonio.
 Gli stereoisomeri configurazionali del tipo mostrato prima necessitano di un prefisso di nomenclatura addizionale da aggiungere al nome IUPAC, allo scopo di specificare le orientazioni spaziali dei gruppi uniti al doppio legame. Sin qui, i prefissi cis- e trans- sono serviti a distinguere gli stereoisomeri; tuttavia, non è sempre chiaro quale isomero dovrebbe essere chiamato cis e quale trans. Per esempio, consideriamo i due composti sotto.
Gli stereoisomeri configurazionali del tipo mostrato prima necessitano di un prefisso di nomenclatura addizionale da aggiungere al nome IUPAC, allo scopo di specificare le orientazioni spaziali dei gruppi uniti al doppio legame. Sin qui, i prefissi cis- e trans- sono serviti a distinguere gli stereoisomeri; tuttavia, non è sempre chiaro quale isomero dovrebbe essere chiamato cis e quale trans. Per esempio, consideriamo i due composti sotto.
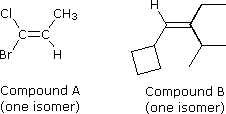 Sia il composto A (1-bromo-1cloropropene) e sia il composto B (1-ciclobutil-2-etil-3-metil-1-butene) possono esistere come una coppia di stereoisomeri configurazionali (è mostrata un solo composto). Come possiamo denominare questi stereoisomeri in modo da specificare la configurazione di ognuno di essi senza ambiguità? L'assegnazione dei prefissi cis e trans ad alcuni di questi stereoisomeri può essere assegnata solo in modo arbitrario, per cui è necessario un metodo più rigoroso. Un sistema completamente privo di ambiguità, basato su di un insieme di regole di priorità dei gruppi, assegna una Z (dal tedesco, zusammen che significa: insieme) o una E (dal tedesco, entgegen che significa: opposto) per designare gli stereoisomeri. Negli isomeri mostrati prima, dove le notazioni cis e trans erano adeguate, Z equivale a cis ed E equivale a trans.
Regola di successione per l'assegnazione delle configurazioni degli alcheni
Stabilire le priorità dei sostituenti sul doppio legame dall'osservazione degli atomi uniti direttamente agli atomi di carbonio del doppio legame.
1. Più è alto il numero atomico dell'atomo diretto sostituente, più è alta la priorità.
Per esempio, H– < C– < N– < O– < Cl– (la priorità aumenta da sinistra a destra).
(A differenti isotopi dello stesso elemento vanno assegnate priorità in accordo con la loro massa atomica.)
2. Se due sostituenti hanno lo stesso atomo diretto sostituente, spostarsi verso l'atomo successivo (allontanandosi dal doppio legame) fino a che si incontri una differenza.
Per esempio, CH3– < C2H5– < ClCH2– < BrCH2– < CH3O–.
Sia il composto A (1-bromo-1cloropropene) e sia il composto B (1-ciclobutil-2-etil-3-metil-1-butene) possono esistere come una coppia di stereoisomeri configurazionali (è mostrata un solo composto). Come possiamo denominare questi stereoisomeri in modo da specificare la configurazione di ognuno di essi senza ambiguità? L'assegnazione dei prefissi cis e trans ad alcuni di questi stereoisomeri può essere assegnata solo in modo arbitrario, per cui è necessario un metodo più rigoroso. Un sistema completamente privo di ambiguità, basato su di un insieme di regole di priorità dei gruppi, assegna una Z (dal tedesco, zusammen che significa: insieme) o una E (dal tedesco, entgegen che significa: opposto) per designare gli stereoisomeri. Negli isomeri mostrati prima, dove le notazioni cis e trans erano adeguate, Z equivale a cis ed E equivale a trans.
Regola di successione per l'assegnazione delle configurazioni degli alcheni
Stabilire le priorità dei sostituenti sul doppio legame dall'osservazione degli atomi uniti direttamente agli atomi di carbonio del doppio legame.
1. Più è alto il numero atomico dell'atomo diretto sostituente, più è alta la priorità.
Per esempio, H– < C– < N– < O– < Cl– (la priorità aumenta da sinistra a destra).
(A differenti isotopi dello stesso elemento vanno assegnate priorità in accordo con la loro massa atomica.)
2. Se due sostituenti hanno lo stesso atomo diretto sostituente, spostarsi verso l'atomo successivo (allontanandosi dal doppio legame) fino a che si incontri una differenza.
Per esempio, CH3– < C2H5– < ClCH2– < BrCH2– < CH3O–.Una volta che le priorità relative dei due sostituenti su ognuno degli atomi di carbonio del doppio legame è stata determinata, si designa con Z l'orientazione cis della coppia a più alta priorità, e con E l'orientazione trans. Applicando queste regole agli isomeri dei composti A e B mostrati sopra, designamo con E la configurazione dell'isomero 1-bromo-1-cloropropene (Br ha priorità più alta di Cl e CH3 più alta di H). La configurazione dell'isomero 1-ciclobutil-2-etil-3-metil-1-butene sarà designata con Z (C4H7 ha piorità più alta di H ed il gruppo isopropile ha priorità più alta di un gruppo etile). L'esempio seguente elabora la determinazione della priorità per un caso più complesso.
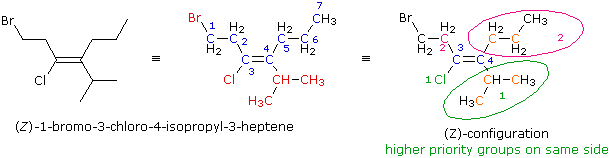 La formula a linee è sviluppata per fornire la formula di struttura osservabile al centro. Il nome di radice è eptene (la catena più lunga che che incorpora entrambi gli atomi di carbonio del doppio legame), ed i sostituenti ad ogni carbonio del doppio legame (in rosso) sono aggiunti per determinare il nome IUPAC. Allo scopo di assegnare un prefisso di configurazione, deve essere determinato l'ordine di priorità dei sostituenti di ogni carbonio del doppi legame. Per il carbonio 3 gli atomi direttamente sostituenti sono un cloro ed un carbonio. Il cloro ha un numero atomico più elevato e quindi ha più alta priorità (colorato in verde e numerato con 1). Il più lontano atomo di bromo non figura in questa scelta. Per il carbonio 4 gli atomi diretti sostituenti sono entrambi atomi di carbonio (colorati in arancione). Come risultato, dobbiamo osservare i successivi atomi con numero atomico più alto nella catena sostituente. Anche questi sono atomi di carbonio, ma il gruppo isopropile ha due atomi di carbonio (anch'essi in arancione) mentre il gruppo propile ne ha solo uno. L'ordine di priorità è quindi: isoprople (verde) > propile (magenta). Dal momento che i due gruppi a più alta priorità (1) sono sullo stesso lato, questa configurazione è Z.
Gli stereoisomeri sono stati osservati anche in certi composti ciclici disostituiti (e multisostituiti). A differenza delle molecole degli alcheni relativamente piatte, le configurazioni dei cicloalcani sostituiti possono essere osservate in tre dimensioni allo scopo di valutare le orientazioni spaziali dei sostituenti. Per convenzione, i chimici utilizzano linee spesse e cuneiformi, per indicare i legami di un sostituente localizzato sopra il piano su cui giace l'anello (notare che i cicloacani costituiti da più di tre atomi di carbonio non sono planari), ed una linea tratteggiata formata da piccoli segmenti paralleli, per indicare i legami di atomi o gruppi localizzati sotto il piano di giacenza dell'anello. Come nel caso degli stereoisomeri del 2-butene, i cicloalcani stereoisomeri disostituiti possono essere designati da prefissi di nomenclatura quali cis e trans. Gli stereoisomeri dell'1,2-dibromociclopentano mostrati sotto ne sono un esempio.
La formula a linee è sviluppata per fornire la formula di struttura osservabile al centro. Il nome di radice è eptene (la catena più lunga che che incorpora entrambi gli atomi di carbonio del doppio legame), ed i sostituenti ad ogni carbonio del doppio legame (in rosso) sono aggiunti per determinare il nome IUPAC. Allo scopo di assegnare un prefisso di configurazione, deve essere determinato l'ordine di priorità dei sostituenti di ogni carbonio del doppi legame. Per il carbonio 3 gli atomi direttamente sostituenti sono un cloro ed un carbonio. Il cloro ha un numero atomico più elevato e quindi ha più alta priorità (colorato in verde e numerato con 1). Il più lontano atomo di bromo non figura in questa scelta. Per il carbonio 4 gli atomi diretti sostituenti sono entrambi atomi di carbonio (colorati in arancione). Come risultato, dobbiamo osservare i successivi atomi con numero atomico più alto nella catena sostituente. Anche questi sono atomi di carbonio, ma il gruppo isopropile ha due atomi di carbonio (anch'essi in arancione) mentre il gruppo propile ne ha solo uno. L'ordine di priorità è quindi: isoprople (verde) > propile (magenta). Dal momento che i due gruppi a più alta priorità (1) sono sullo stesso lato, questa configurazione è Z.
Gli stereoisomeri sono stati osservati anche in certi composti ciclici disostituiti (e multisostituiti). A differenza delle molecole degli alcheni relativamente piatte, le configurazioni dei cicloalcani sostituiti possono essere osservate in tre dimensioni allo scopo di valutare le orientazioni spaziali dei sostituenti. Per convenzione, i chimici utilizzano linee spesse e cuneiformi, per indicare i legami di un sostituente localizzato sopra il piano su cui giace l'anello (notare che i cicloacani costituiti da più di tre atomi di carbonio non sono planari), ed una linea tratteggiata formata da piccoli segmenti paralleli, per indicare i legami di atomi o gruppi localizzati sotto il piano di giacenza dell'anello. Come nel caso degli stereoisomeri del 2-butene, i cicloalcani stereoisomeri disostituiti possono essere designati da prefissi di nomenclatura quali cis e trans. Gli stereoisomeri dell'1,2-dibromociclopentano mostrati sotto ne sono un esempio.
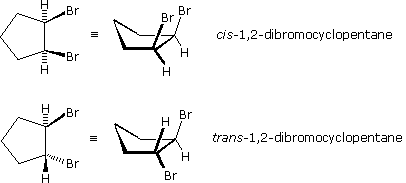
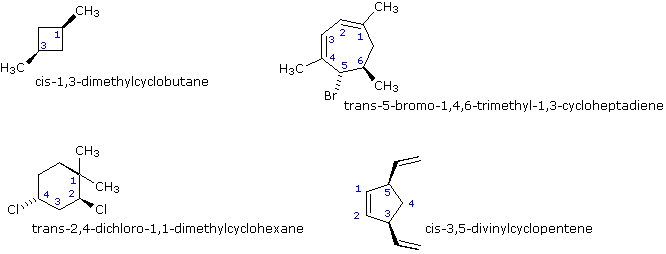
In generale, se in un anello due atomi di carbonio sp3 hanno due differenti gruppi sostituenti (senza tener conto di altri atomi dell'anello), lo stereoisomerismo è possibile. Questo è simile all'esempio di sostituzione che causa la stereoisomeria negli alcheni; invero, si potrebbe guardare ad un doppio legame come ad un anello a due membri. Altri quattro esempi di questo genere di stereoisomerismo nei composti ciclici sono mostrati sotto.
 Se più di due atomi di carbonio dell'anello hanno sostituenti differenti (senza tenere in conto altri atomi dell'anello) la notazione stereochimica che distingue i vari isomeri diventa più complessa.
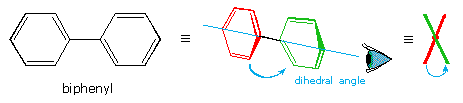
Le formule di struttura mostrano il modo in cui gli atomi della molecola sono legati insieme (la sua costituzione), ma generalmente non descrivono la forma tridimensionale di una molecola, a meno che non siano utilizzate speciali notazioni per i legami (ad es. linee cuneiformi o tratteggiate). L'importanza che tali formule possano descrivere la struttura tridimensiomnale diventa chiara studiando lo stereoisomerismo configurazionale, dove l'orientazione relativa degli atomi nello spazio è fissata dalla costituzione dei legami di una molecola (ad es. doppi legami ed anelli). E' stato anche sottolineato che i prefissi di nomenclatura possono essere utilizzati nella denominazione di stereoisomeri specifici. In questo capitolo estenderemo la nostra visione tridimensionale della struttura molecolare per includere composti che normalmente assumono un insieme di orientazioni spaziali tridimensionali che caratterizzano lo stesso composto isolabile. Noi chiamiamo queste differenti orientazioni spaziali degli atomi di una molecola che risultano da rotazioni o da torsioni di singoli legami, conformazioni.
Se più di due atomi di carbonio dell'anello hanno sostituenti differenti (senza tenere in conto altri atomi dell'anello) la notazione stereochimica che distingue i vari isomeri diventa più complessa.
Le formule di struttura mostrano il modo in cui gli atomi della molecola sono legati insieme (la sua costituzione), ma generalmente non descrivono la forma tridimensionale di una molecola, a meno che non siano utilizzate speciali notazioni per i legami (ad es. linee cuneiformi o tratteggiate). L'importanza che tali formule possano descrivere la struttura tridimensiomnale diventa chiara studiando lo stereoisomerismo configurazionale, dove l'orientazione relativa degli atomi nello spazio è fissata dalla costituzione dei legami di una molecola (ad es. doppi legami ed anelli). E' stato anche sottolineato che i prefissi di nomenclatura possono essere utilizzati nella denominazione di stereoisomeri specifici. In questo capitolo estenderemo la nostra visione tridimensionale della struttura molecolare per includere composti che normalmente assumono un insieme di orientazioni spaziali tridimensionali che caratterizzano lo stesso composto isolabile. Noi chiamiamo queste differenti orientazioni spaziali degli atomi di una molecola che risultano da rotazioni o da torsioni di singoli legami, conformazioni.Nel caso dell'esano, abbiamo una catena non ramificata di sei atomi di carbonio che è scritta molto spesso con una formula lineare: CH3CH2CH2CH2CH2CH3. Noi sappiamo che ciò non è strettamente vero, dal momento che gli atomi di carbonio hanno tutti una configurazione tetraedrica. La reale forma della catena estesa è quindi in natura a zig-zag. Tuttavia, avviene un'agevole rotazione intorno ai legami carbonio-carbonio, e la catena a sei atomi di carbonio si avvolge facilmente per dare una forma abbastanza differente. Molte conformazioni dell'esano sono possibili. Il semplice alcano etano è adatto per una buona introduzione all'analisi conformazionale. Qui vi è un solo legame carbonio - carbonio, e la strutture rotazionali (rotameri) che esso può assumere cadono tra due estremi, sfalsata ed eclissata. Nella seguente descrizione di questi conformeri, sono usate diverse notazioni strutturali. La prima vede la molecola dell'etano lateralmente, con il legame carbonio-carbonio in posizione orizzontale rispetto all'osservatore. Gli atomi d'idrogeno vengono allora localizzati nello spazio circostante da legami cuneiformi (frontalmente al piano) e tratteggiati (posteriormente al piano). Se si ruota questa struttura in modo che il carbonio 1 sia inclinato verso il basso e portato più vicino all'osservatore, si presenta la proiezione a "cavalletto da falegname". Infine, se l'osservatore guarda dall'alto in basso il legame carbonio - carbonio con il carbonio 1 di fronte al carbonio 2, vede la proiezione di Newman.
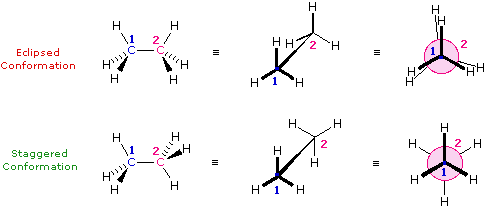
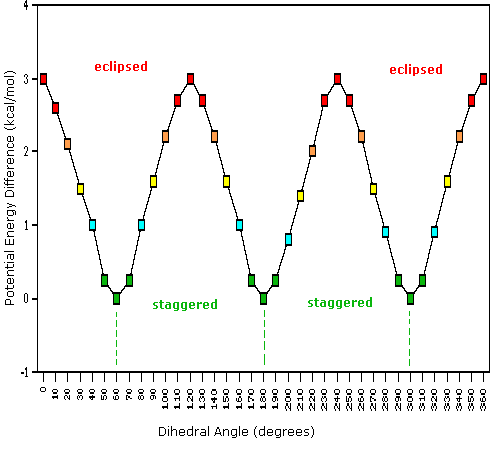
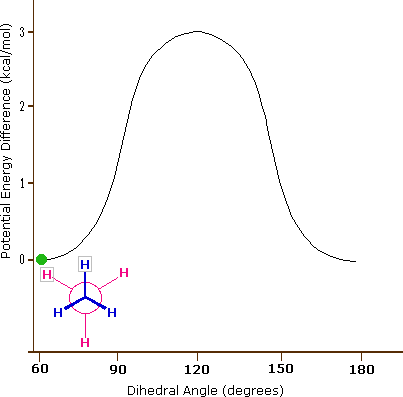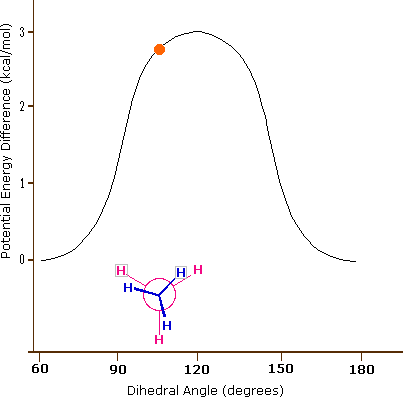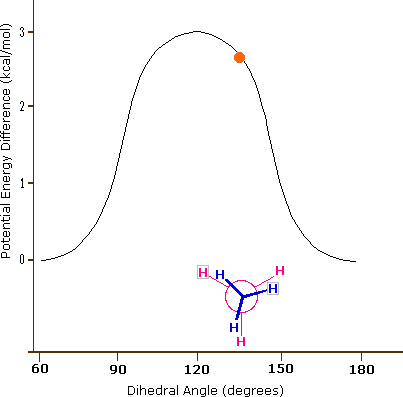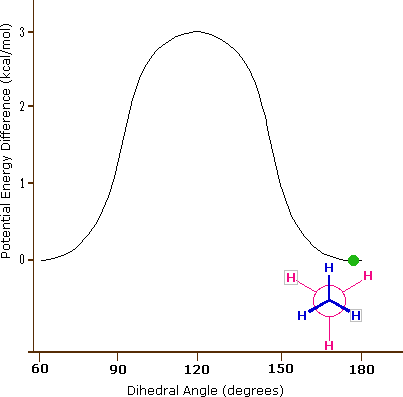
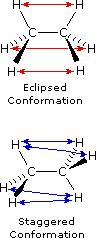 Come risultato delle repulsioni legame-elettrone, illustrate sopra a destra, la conformazione eclissata è meno stabile di quella sfalsata di circa 3 kcal/mole (tensione di eclissamento). Le più intense repulsioni nella conformazione eclissata sono rappresentate da frecce rosse. Vi sono altre sei repulsioni meno forti che non sono mostrate. Nella conformazione sfalsata ci sono sei repulsioni uguali di legame, quattro delle quali sono rappresentate da frecce blu, e queste sono tutte sostanzialmente meno intense delle tre repulsioni più forti della conformazione eclissata. Di conseguenza, l'energia potenziale associata con le diverse conformazioni dell'etano varia con l'angolo diedro dei legami, come mostrato sotto. Sebbene i conformeri dell'etano siano in rapido equilibrio l'un con l'altro, la differenza di energia di 3 kcal/mole porta ad una sostanziale preponderanza dei conformeri sfalsati (> 99,9 %) in ogni dato momento.
Come risultato delle repulsioni legame-elettrone, illustrate sopra a destra, la conformazione eclissata è meno stabile di quella sfalsata di circa 3 kcal/mole (tensione di eclissamento). Le più intense repulsioni nella conformazione eclissata sono rappresentate da frecce rosse. Vi sono altre sei repulsioni meno forti che non sono mostrate. Nella conformazione sfalsata ci sono sei repulsioni uguali di legame, quattro delle quali sono rappresentate da frecce blu, e queste sono tutte sostanzialmente meno intense delle tre repulsioni più forti della conformazione eclissata. Di conseguenza, l'energia potenziale associata con le diverse conformazioni dell'etano varia con l'angolo diedro dei legami, come mostrato sotto. Sebbene i conformeri dell'etano siano in rapido equilibrio l'un con l'altro, la differenza di energia di 3 kcal/mole porta ad una sostanziale preponderanza dei conformeri sfalsati (> 99,9 %) in ogni dato momento.
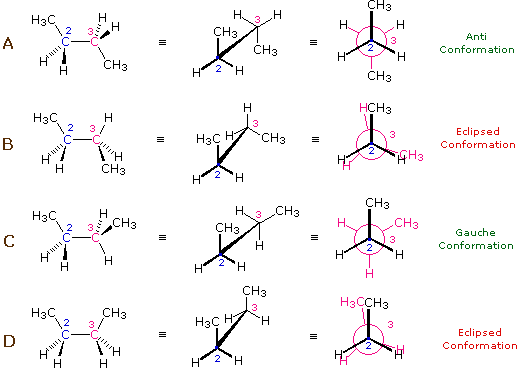
 L'idrocarburo butano ha un insieme di conformazioni associate con la sua costituzione più grande e complesso dell'etano. Particolarmente interessanti ed importanti sono le conformazioni prodotte per rotazione intorno al legame centrale carbonio - carbonio. Tra queste, focalizzeremo la nostra attenzione su due conformeri sfalsati (A e C) e due conformeri eclissati (B e D), mostrate sotto in diverse rappresentazioni stereoscopiche. Come nel caso dell'etano, i conformeri sfalsati sono più stabili di quelli eclissati tra 2,8 e 4,5 kcal/mole. Dal momento che i conformeri sfalsati rappresentano i componenti principali di un campione di butano essi sono designati dai prefissi anti per A e gauche per C.
L'idrocarburo butano ha un insieme di conformazioni associate con la sua costituzione più grande e complesso dell'etano. Particolarmente interessanti ed importanti sono le conformazioni prodotte per rotazione intorno al legame centrale carbonio - carbonio. Tra queste, focalizzeremo la nostra attenzione su due conformeri sfalsati (A e C) e due conformeri eclissati (B e D), mostrate sotto in diverse rappresentazioni stereoscopiche. Come nel caso dell'etano, i conformeri sfalsati sono più stabili di quelli eclissati tra 2,8 e 4,5 kcal/mole. Dal momento che i conformeri sfalsati rappresentano i componenti principali di un campione di butano essi sono designati dai prefissi anti per A e gauche per C.
 A questo punto risulta utile riassumere alcuni aspetti importanti dello stereoisomerismo conformazionale.
(i) La maggior parte delle interconversioni conformazionali in molecole semplici avvengono rapidamente a temperatura ambiente. Di conseguenza, l'isolamento di conformeri puri non è normalmente possibile.
(ii) Conformeri specifici richiedono una speciale terminologia di nomenclatura con termini quali sfalsato, eclissato, gauche e anti, quando devono essere designati.
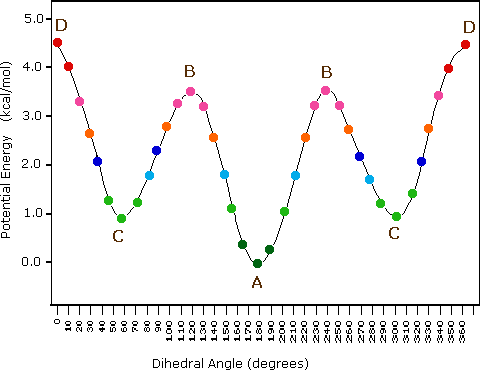
(iii) Conformeri specifici possono anche essere designati dagli angoli diedri. Nei conformari del butano mostrati sopra, gli angoli diedri formati dai due gruppi metilici intorno al doppio legame centrale sono: A 180º, B 120º, C 60º e D 0º.
(iv) Le conformazioni sfalsate intorno ai legame singoli carbonio-carbonio sono più stabili (hanno una più bassa energia potenziale) delle corrispondenti conformazioni eclissate. La più alta energia dei legami eclissati è conosciuta come tensione di eclissamento.
(v) Nel butano, il conformero gauche è meno stabile del conformero anti di circa 0,9 kcal/mole. Ciò è dovuto ad un affolamento dei due gruppi metilici nella struttura gauche, ed è chiamata tensione sterica o intralcio sterico.
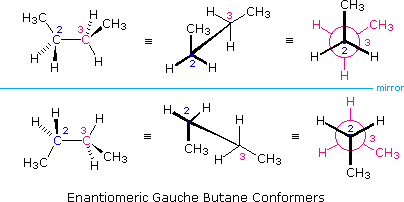
(vi) I conformeri B e C del butano non hanno strutture identiche per riflessione in quanto gli angoli diedri, misurati in senso orario, sono rispettivamente di 240º e 300º. Queste coppie sono uguali dal punto di vista energetico, e non sono state distinte nel diagramma di energia qui mostrato.
A questo punto risulta utile riassumere alcuni aspetti importanti dello stereoisomerismo conformazionale.
(i) La maggior parte delle interconversioni conformazionali in molecole semplici avvengono rapidamente a temperatura ambiente. Di conseguenza, l'isolamento di conformeri puri non è normalmente possibile.
(ii) Conformeri specifici richiedono una speciale terminologia di nomenclatura con termini quali sfalsato, eclissato, gauche e anti, quando devono essere designati.
(iii) Conformeri specifici possono anche essere designati dagli angoli diedri. Nei conformari del butano mostrati sopra, gli angoli diedri formati dai due gruppi metilici intorno al doppio legame centrale sono: A 180º, B 120º, C 60º e D 0º.
(iv) Le conformazioni sfalsate intorno ai legame singoli carbonio-carbonio sono più stabili (hanno una più bassa energia potenziale) delle corrispondenti conformazioni eclissate. La più alta energia dei legami eclissati è conosciuta come tensione di eclissamento.
(v) Nel butano, il conformero gauche è meno stabile del conformero anti di circa 0,9 kcal/mole. Ciò è dovuto ad un affolamento dei due gruppi metilici nella struttura gauche, ed è chiamata tensione sterica o intralcio sterico.
(vi) I conformeri B e C del butano non hanno strutture identiche per riflessione in quanto gli angoli diedri, misurati in senso orario, sono rispettivamente di 240º e 300º. Queste coppie sono uguali dal punto di vista energetico, e non sono state distinte nel diagramma di energia qui mostrato.Sebbene la rappresentazione abituale con l'uso di linee per i semplici cicloalcani diano poligoni geometrici, la reale forma di questi composti è molto diversa nella maggioranza dei casi. Il ciclopropano è necessariamente planare (piatto), con gli atomi di carbonio posti agli angoli di un triangolo equilatero. Gli angoli di legame di 60º sono molto più piccoli degli angoli ottimali a 109,5º di un normale atomo di carbonio tetraedrico, ed la risultante tensione angolare influenza drammaticamente il comportamento chimico di questo cicloalcano. Il ciclopropano soffre anche della tensione di eclissamento, dal momento che tutti i legami carbonio-carbonio sono completamente eclissati. Il ciclobutano riduce una certa parte della tensione di eclissamento sui legami mediante ripiegamento (l'angolo diedro formato con il piano è di circa 25º), ma l'eclissamento totale e la tensione angolare resta alta. Il ciclopentano ha una tensione angolare molto ridotta (gli angoli di un pentagono sono di 108º), ma la sua tensione di eclissamento sarebbe elevata (circa 10 kcal/mole) se esso rimanesse planare. Di conseguenza, l'anello a cinque membri adotta una conformazione non planare ripiegata ogni volta che sia possibile. Anelli più grandi di quello del ciclopentano avrebbero tensioni angolari se fossero planari. Tuttavia, questa questa tensione, insieme con la tensione di eclissamento intrinseca in un struttura planare, può essere mitigata dal ripiegamento dell'anello. Il cicloesano è un buon esempio di un sistema ciclico del carbonio che elimina virtualmente tensione angolare e di eclissamento dall'adozione di conformazioni non planari, tali come quelle mostrati sotto. Il cicloeptano ed il cicloottano hanno tensioni più elevate del cicloesano, in larga parte dovute ad affollamento transanulare (impedimento sterico dovuto a gruppi posti sui lati opposti dell'anello). Alcune conformazioni degli anelli del cicloesano
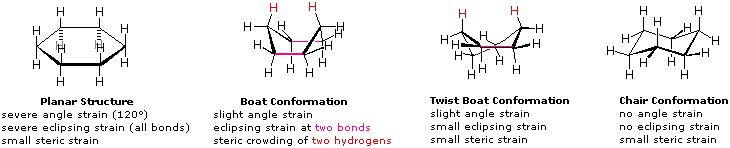 Una struttura planare del cicloesano è chiaramente improbabile. Gli angoli di legame sarebbero necessariamente di 120º, cioè di 10,5º più grandi dell'angolo tetraedrico ideale. Ancora, ogni legame carbonio-carbonio in una tale struttura sarebbe eclissato. L'angolo risultante e le tensioni di eclissamento destabilizzerebbero severamente questa struttura. Se due atomi di carbonio sui lati opposti dell'anello a sei membri si sollevano rispetto al piano dell'anello, molta della tensione angolare può essere eliminata. Questa struttura a barca possiede ancora due legami eclissati (di colore magenta nel disegno) ed un intenso affollamento sterico dei due atomi d'idrogeno sulla "prua" e sulla "poppa" della barca. L'affollamento sterico è spesso chiamato impedimento sterico. Mediante torsione della conformazione a barca, l'impedimento sterico può essere parzialmente ridotto, ma questa conformazione a barca ritorta conserva ancora una certa parte delle tensioni caratteristiche della conformazione a barca. Infine, sollevando un atomo di carbonio al di sopra del piano dell'anello e l'altro abbassandolo sotto il piano, si ottiene una conformazione a sedia relativamente libera da tensioni. Questa è la struttura predominante adottata dalla molecola del cicloesano.
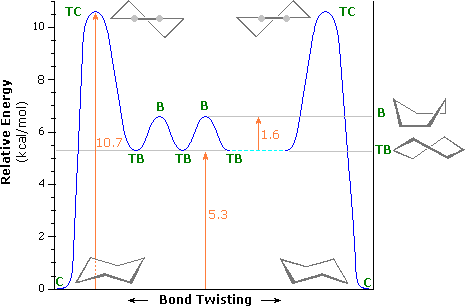
Un diagramma di energia di interconversione tra queste conformazioni è riportato sotto. L'energia di attivazione per la conversione barca-barca è dovuta principalmente alla più alta energia della forma a sedia ritorta (TC, Twist-Chair), in cui sono presenti tensioni angolari e di eclissamento significative. Un agevole equilibrio tra barca ritorta (TB, Twist Boat) e barca (B, Boat) ha luogo così come un conformero a sedia (C, Chair) si trasforma nell'altro.
Una struttura planare del cicloesano è chiaramente improbabile. Gli angoli di legame sarebbero necessariamente di 120º, cioè di 10,5º più grandi dell'angolo tetraedrico ideale. Ancora, ogni legame carbonio-carbonio in una tale struttura sarebbe eclissato. L'angolo risultante e le tensioni di eclissamento destabilizzerebbero severamente questa struttura. Se due atomi di carbonio sui lati opposti dell'anello a sei membri si sollevano rispetto al piano dell'anello, molta della tensione angolare può essere eliminata. Questa struttura a barca possiede ancora due legami eclissati (di colore magenta nel disegno) ed un intenso affollamento sterico dei due atomi d'idrogeno sulla "prua" e sulla "poppa" della barca. L'affollamento sterico è spesso chiamato impedimento sterico. Mediante torsione della conformazione a barca, l'impedimento sterico può essere parzialmente ridotto, ma questa conformazione a barca ritorta conserva ancora una certa parte delle tensioni caratteristiche della conformazione a barca. Infine, sollevando un atomo di carbonio al di sopra del piano dell'anello e l'altro abbassandolo sotto il piano, si ottiene una conformazione a sedia relativamente libera da tensioni. Questa è la struttura predominante adottata dalla molecola del cicloesano.
Un diagramma di energia di interconversione tra queste conformazioni è riportato sotto. L'energia di attivazione per la conversione barca-barca è dovuta principalmente alla più alta energia della forma a sedia ritorta (TC, Twist-Chair), in cui sono presenti tensioni angolari e di eclissamento significative. Un agevole equilibrio tra barca ritorta (TB, Twist Boat) e barca (B, Boat) ha luogo così come un conformero a sedia (C, Chair) si trasforma nell'altro.
 Gli studi riguardanti le conformazioni dell'esano furono iniziati da H. Sachse (1890) e E. Mohr (1918), ma essi non furono, fino al 1950, che un completo trattamento delle conseguenze del ripiegamento di interconversione tra i conformeri a sedia e le differenti orientazioni dei legami sospesi furono delucidate da D. H. R. Barton (Premio Nobel 1969 insieme a O. Hassel). La seguente discussione presenta alcune delle caratteristiche essenziali dell'analisi conformazionale.
Dopo un accurato esame di una conformazione a sedia del cicloesano, troviamo che i dodici atomi di idrogeno non sono strutturalmente equivalenti. Sei atomi sono localizzati intorno alla periferia dell'anello di carbonio, e sono chiamati equatoriali. Gli altri sei sono orientati sopra e sotto l'approssimativo piano dell'anello (tre per ognuna delle posizioni), e sono chiamati assiali poiché essi sono allineati parallelamente all'asse di simmetria dell'anello. Nel modello ad aste mostrato in basso a sinistra, gli idrogeni equatoriali sono colorati in blu, e quelli assiali in rosso. Poiché vi sono due conformazioni a sedia equivalenti del cicloesano in rapido equilibrio, tutti i dodici atomi d'idrogeno hanno un carattere del 50% equatoriale e del 50% assiale.
Gli studi riguardanti le conformazioni dell'esano furono iniziati da H. Sachse (1890) e E. Mohr (1918), ma essi non furono, fino al 1950, che un completo trattamento delle conseguenze del ripiegamento di interconversione tra i conformeri a sedia e le differenti orientazioni dei legami sospesi furono delucidate da D. H. R. Barton (Premio Nobel 1969 insieme a O. Hassel). La seguente discussione presenta alcune delle caratteristiche essenziali dell'analisi conformazionale.
Dopo un accurato esame di una conformazione a sedia del cicloesano, troviamo che i dodici atomi di idrogeno non sono strutturalmente equivalenti. Sei atomi sono localizzati intorno alla periferia dell'anello di carbonio, e sono chiamati equatoriali. Gli altri sei sono orientati sopra e sotto l'approssimativo piano dell'anello (tre per ognuna delle posizioni), e sono chiamati assiali poiché essi sono allineati parallelamente all'asse di simmetria dell'anello. Nel modello ad aste mostrato in basso a sinistra, gli idrogeni equatoriali sono colorati in blu, e quelli assiali in rosso. Poiché vi sono due conformazioni a sedia equivalenti del cicloesano in rapido equilibrio, tutti i dodici atomi d'idrogeno hanno un carattere del 50% equatoriale e del 50% assiale.
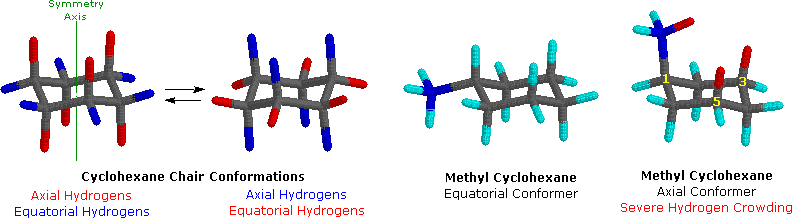
Poiché i legami assiali sono paralleli tra di loro, i sostituenti più grandi dell'idrogeno generalmente soffrono di un affollamento sterico più elevato quando essi sono orientati in modo assiale piuttosto che equatoriale. Di conseguenza, i cicloesani adotteranno preferenzialmente conformazioni in cui i sostituenti più grandi assumono orientazione equatoriale. Nei due conformeri del metilcicloesano mostrati sopra, il carbonio del metile è colorato in blu. Quando il gruppo metile occupa una posizione assiale esso soffre di affollamento sterico dovuto ai due atomi d'idrogeno assiali localizzati sullo stesso lato dell'anello. Questo affollamento o impedimento sterico è associato con gli atomi d'idrogeno della struttura colorati in rosso. Un attento esame del conformero assiale mostra che questo impedimento sterico a due orientazioni gauche-simili del gruppo metile con gli atomi di carbonio 3 e 5 dell'anello. L'uso di modelli, incluso Chime, risulta particolarmente utile nella individuazione e nella valutazione di queste associazioni. Il relativo impedimento sterico provato da differenti gruppi sostituenti orientati in una posizione assiale rispetto ad una equatoriale può essere determinata dall'equilibrio conformazionale del composto. La corrispondente costante di equilibrio è in relazione con la differenza di energia tra i conformeri, e la raccolta di tali dati ci permette di valutare la relativa tendenza dei sostituenti ad esistere in una posizione equatoriale o assiale. È chiaro che la "dimensione" apparente di un sostituente è influenzata dall'ampiezza e dalla lunghezza del suo legame con il cicloesano, come si evidenzia dal fatto che un gruppo vinilico assiale è meno impedito dell'etile e lo iodio leggermente meno del cloro.
Dal momento che il cicloesano è così comune tra i composti naturali e sintetici, e poichè le sue proprietà conformazionali sono conosciute abbastanza bene, in questa discussione focalizzeremo la nostra attenzione sull'anello a sei membri del cicloesano. In un campione di cicloesano, i due identici conformeri a sedia sono presenti in eguale concentrazione e gli atomi d'idrogeno sono tutti equivalenti (50% equatoriali e 50% assiali), fatto dovuto alla rapida interconversione dei conformeri. Quando l'anello del cicloesano reca con sé un sostituente, i due conformeri a sedia non sono gli stessi. In un conformero il sostituente è assiale, nell'altro è equatoriale. A causa dell'impedimento sterico nella posizione assiale, i gruppi sostituenti preferiscono assumere posizioni equatoriali diventando predominati nell'equilibrio. Abbiamo prima notato che i cicloalcani aventi due o più sostituenti su atomi di carbonio differenti dell'anello, essi esistono come coppie (qualche volta più di due) di stereoisomeri configurazionali. Dobbiamo ora esaminare il modo con cui conformazioni favorite dell'anello influenzano le proprietà degli isomeri configurazionali. Ricordare che gli stereoisomeri configurazionali sono stabili e non facilmente interconvertibili, mentre gli isomeri conformazionali normalmente s'interconvertono rapidamente. Nello studiare possibili strutture dei cicloesani sostituiti, risulta utile seguire due principi. (i) Le conformazioni a sedia sono generalmente più stabili delle altre possibili. (ii) I sostituenti sui conformeri a sedia preferiscono occupare posizioni equatoriali in quanto le posizioni assiali aumentano l'impedimento sterico. Le seguenti equazioni e formule illustrano come la presenza di due o più sostituenti su di un anello di cicloesano perturba l'interconversione dei due conformeri a sedia in modalità che possono essere del tutto previste. Strutture conformazionali di cicloesani disostituiti 1,1-dimetilcicloesano
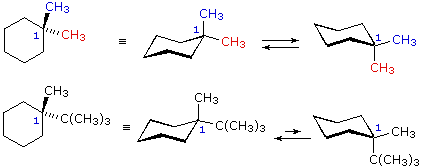 1-t-butil-1-metilcicloesano
cis-1,2-dimetilcicloesano
1-t-butil-1-metilcicloesano
cis-1,2-dimetilcicloesano 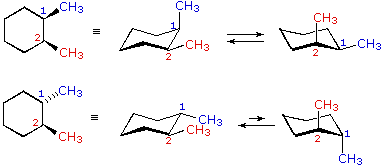 trans-1,2-dimetilcicloesano
cis-1,3-dimetilcicloesano
trans-1,2-dimetilcicloesano
cis-1,3-dimetilcicloesano 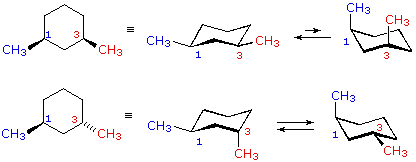 trans-1,3-dimetilcicloesano
cis-1,4-dimetilcicloesano
trans-1,3-dimetilcicloesano
cis-1,4-dimetilcicloesano 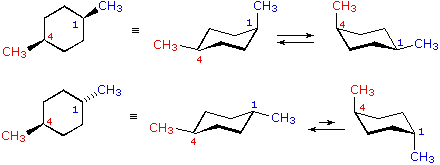 trans-1,4-dimetilcicloesano
Nel caso dei cicloesani 1,1-disostituiti, uno dei sostituenti deve essere necessariamente assiale e l'altro equatoriale, senza tener conto di quale conformero a sedia si stia considerando. Dal momento che i sostituenti nel 1,1-dimetilcicloesano sono gli stessi, i due conformeri sono identici e presenti in egual concentrazione, Nell' 1-t-butil-1-metilcicloesano il gruppo t-butile è molto più grande del metile, e quel conformero a sedia in cui il gruppo più grande è equatoriale sarà favorito nell'equilibrio (> 99%). Conseguentemente, il gruppo metile in questo composto è quasi esclusivamente assiale nella sua orientazione.
Nei casi dei composti disostituiti 1,2-, 1,3- e 1,4-, l'analisi è un po' più complessa. È sempre possibile avere entrambi i gruppi equatoriali, ma se ciò richiede una relazione cis o trans, allora dipende dalla posizione relativa dei sostituenti. Se noi contiamo attorno all'anello dal carbonio1 al carbonio 6, il legame con numero più elevato su ogni carbonio cambia la sua orientazione da equatoriale (o assiale) ad assiale (o equatoriale) e viceversa. È importante ricordare che i legami in una data posizione di un conformero anulare a sedia si alternano sempre in questa maniera. Quindi, dovrebbe essere chiaro per per i composti cis-1,2 disostituiti, uno dei sostituenti deve essere equatoriale e l'altro assiale; nell'isomero trans entrambi possono essere equatoriali. A causa della natura alternante dei legami equatoriali ed assiali, la relazione opposta è vera per i composti 1,3-disostituiti (cis è completamente equatoriale e trans è sia equatoriale che assiale). Infine, i composti 1,4-disostituiti si comportano come i composti 1,2-disostituiti.
trans-1,4-dimetilcicloesano
Nel caso dei cicloesani 1,1-disostituiti, uno dei sostituenti deve essere necessariamente assiale e l'altro equatoriale, senza tener conto di quale conformero a sedia si stia considerando. Dal momento che i sostituenti nel 1,1-dimetilcicloesano sono gli stessi, i due conformeri sono identici e presenti in egual concentrazione, Nell' 1-t-butil-1-metilcicloesano il gruppo t-butile è molto più grande del metile, e quel conformero a sedia in cui il gruppo più grande è equatoriale sarà favorito nell'equilibrio (> 99%). Conseguentemente, il gruppo metile in questo composto è quasi esclusivamente assiale nella sua orientazione.
Nei casi dei composti disostituiti 1,2-, 1,3- e 1,4-, l'analisi è un po' più complessa. È sempre possibile avere entrambi i gruppi equatoriali, ma se ciò richiede una relazione cis o trans, allora dipende dalla posizione relativa dei sostituenti. Se noi contiamo attorno all'anello dal carbonio1 al carbonio 6, il legame con numero più elevato su ogni carbonio cambia la sua orientazione da equatoriale (o assiale) ad assiale (o equatoriale) e viceversa. È importante ricordare che i legami in una data posizione di un conformero anulare a sedia si alternano sempre in questa maniera. Quindi, dovrebbe essere chiaro per per i composti cis-1,2 disostituiti, uno dei sostituenti deve essere equatoriale e l'altro assiale; nell'isomero trans entrambi possono essere equatoriali. A causa della natura alternante dei legami equatoriali ed assiali, la relazione opposta è vera per i composti 1,3-disostituiti (cis è completamente equatoriale e trans è sia equatoriale che assiale). Infine, i composti 1,4-disostituiti si comportano come i composti 1,2-disostituiti.
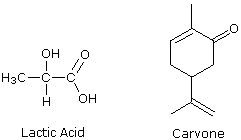
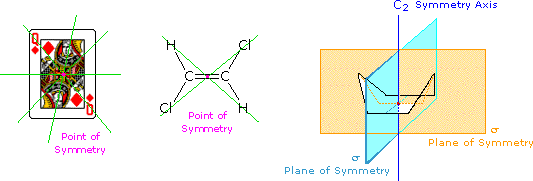
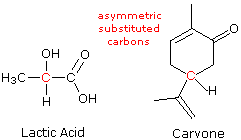
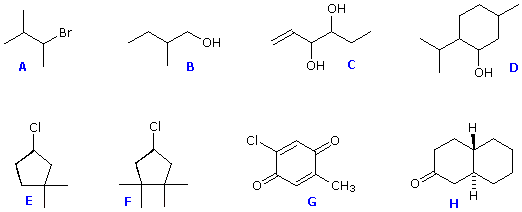
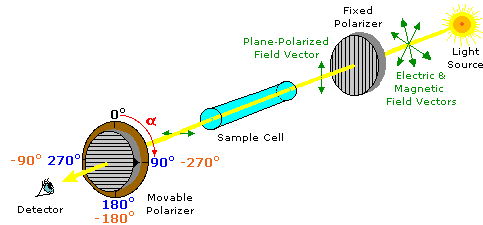
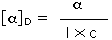 ove l = lunghezza di cella in dm, c = concentrazione in g/ml e D è la luce a 589 nm da una lampada al sodio
ove l = lunghezza di cella in dm, c = concentrazione in g/ml e D è la luce a 589 nm da una lampada al sodio
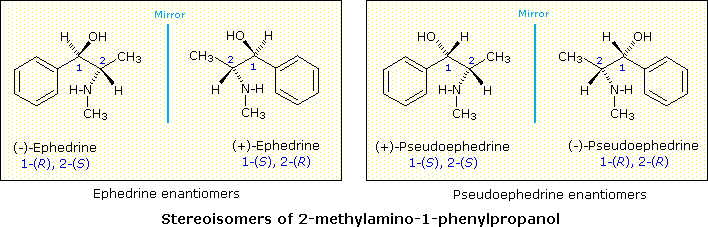
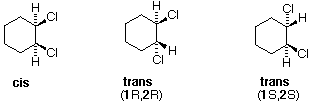
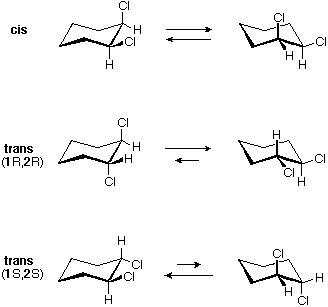
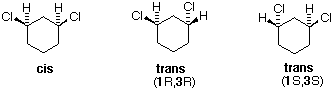
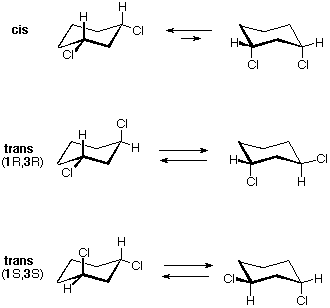 Tutti gli isomeri 1,2-dicloro sono isomeri costituzionali degli isomeri 1,3-dicloro. In ognuna delle categorie (1,2- e 1,3-), l'isomero (R,R)-trans e l'isomero (S,S)-trans sono enantiomeri. L'isomero cis- è un diastereoisomero degli isomeri trans-. Infine, tutti questi isomeri possono esistere come miscela di due (o più) isomeri conformazionali, come illustrato nella tavola.
Il conformero a sedia dell'isomero cis 1,2-dicloro è chirale. Esso esiste come miscela 50:50 di conformazioni enantiomeriche, che s'interconvertono così rapidamente da non poter essere risolti (cioè, separati). Dal momento che l'isomero cis ha due centri di chiralità (atomi di carbonio asimmetrici) ed è inattivo otticamente, esso è un meso composto. I corrispondenti isomeri trans possono anche esistere sotto forma di conformazioni chirali rapidamente interconvertenti. Il conformero diequatoriale predomina in ogni caso, con le conformazioni (R,R) che sono immagini speculari di quelle (S,S). Tutte queste conformazioni sono diastereoisomeriche con conformazioni cis.
Il conformero diequatoriale a sedia dell'isomero cis-1,3-dicloro è chirale. Esso è il maggior componente di un veloce equilibrio con il conformero diassiale, anch'esso chirale. Anche questo isomero è un meso composto. Pure il corrispondente isomero trans è sottoposto ad una rapida interconversione, per cui ogni enantiomero (R,R) e (S,S) ha predominantemente una singola conformazione chirale. Queste conformazioni enantiomeriche sono diastereomeriche con le conformazioni cis.
L' 1,4-diclorocicloesano può esistere come stereoisomero trans o cis. Entrambi sono chirali, poiché gli anelli a sei membri disostituiti hanno un piano di simmetria. Questi isomeri sono diastereoisomeri l'un dell'altro, e sono isomeri costituzionali degli isomeri 1,2- e 1,3-.
Tutti gli isomeri 1,2-dicloro sono isomeri costituzionali degli isomeri 1,3-dicloro. In ognuna delle categorie (1,2- e 1,3-), l'isomero (R,R)-trans e l'isomero (S,S)-trans sono enantiomeri. L'isomero cis- è un diastereoisomero degli isomeri trans-. Infine, tutti questi isomeri possono esistere come miscela di due (o più) isomeri conformazionali, come illustrato nella tavola.
Il conformero a sedia dell'isomero cis 1,2-dicloro è chirale. Esso esiste come miscela 50:50 di conformazioni enantiomeriche, che s'interconvertono così rapidamente da non poter essere risolti (cioè, separati). Dal momento che l'isomero cis ha due centri di chiralità (atomi di carbonio asimmetrici) ed è inattivo otticamente, esso è un meso composto. I corrispondenti isomeri trans possono anche esistere sotto forma di conformazioni chirali rapidamente interconvertenti. Il conformero diequatoriale predomina in ogni caso, con le conformazioni (R,R) che sono immagini speculari di quelle (S,S). Tutte queste conformazioni sono diastereoisomeriche con conformazioni cis.
Il conformero diequatoriale a sedia dell'isomero cis-1,3-dicloro è chirale. Esso è il maggior componente di un veloce equilibrio con il conformero diassiale, anch'esso chirale. Anche questo isomero è un meso composto. Pure il corrispondente isomero trans è sottoposto ad una rapida interconversione, per cui ogni enantiomero (R,R) e (S,S) ha predominantemente una singola conformazione chirale. Queste conformazioni enantiomeriche sono diastereomeriche con le conformazioni cis.
L' 1,4-diclorocicloesano può esistere come stereoisomero trans o cis. Entrambi sono chirali, poiché gli anelli a sei membri disostituiti hanno un piano di simmetria. Questi isomeri sono diastereoisomeri l'un dell'altro, e sono isomeri costituzionali degli isomeri 1,2- e 1,3-.
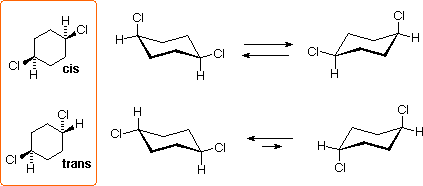 Tutti i conformeri a sedia di questi isomeri sono chirali, ed il conformero equatoriale dell'isomero trans è la specie predomeinante all'equilibrio.
Tutti i conformeri a sedia di questi isomeri sono chirali, ed il conformero equatoriale dell'isomero trans è la specie predomeinante all'equilibrio.
 Ci aggiorneremo per ampliare questa pagina, per il momento buona lettura
Ci aggiorneremo per ampliare questa pagina, per il momento buona lettura