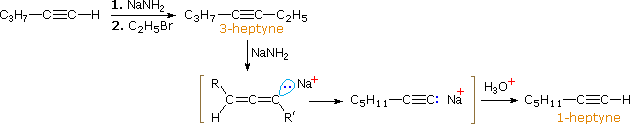quimico
2011-12-08 19:21
Reazioni di addizione
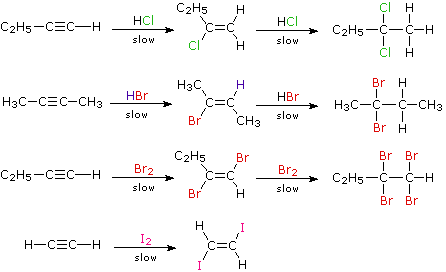
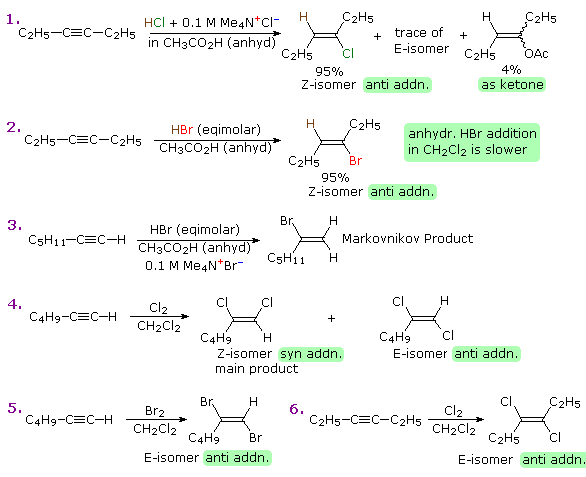
Un triplo legame carbonio-carbonio può essere situato in qualsiasi posizione non ramificata con una catena carboniosa o all'estremità di una catena, in tale caso si dice terminale. A causa della sua configurazione lineare (l'angolo di legame di un carbonio ibridizzato sp è di 180°), un anello di carbonio a dieci termini è il più piccolo che possa accomodare questa funzione senza eccessiva tensione. Dato che la più comune trasformazione chimica di un doppio legame carbonio-carbonio è una reazione di addizione, ci dovremmo aspettare che lo stesso sia vero per i tripli legami carbonio-carbonio. Inoltre, la maggior parte delle reazioni di addizione di alcheni avvengono anche con gli alchini, e con una regio- e stereoselettività simile.
1. Idrogenazione catalitica
L'addizione catalitica dell'idrogeno al 2-butino non solo serve come esempio di una tale reazione di addizione, ma anche fornisce i dati sul calore di reazione che riflette le stabilità termodinamiche relative di questi idrocarburi, come mostrato nello schema sotto.
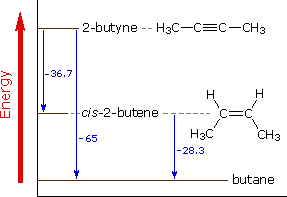
Dai calori di idrogenazione, mostrati in blu in kcal/mole, dovrebbe apparire che gli alchini sono termodinamicamente meno stabili degli alcheni in un modo maggiore rispetto a quanto gli alcheni sono meno stabili degli alcani. Le energia di legame standard per i legami carbonio-carbonio confermano questa conclusione. Perciò, un doppio legame è più forte di un legame singolo, ma non è forte il doppio. La differenza (63 kcal/mole) può essere considerata come la forza della componente legame π. In modo simile, un triplo legame è più forte di un doppio legame, ma non è più forte del 50%. Qui la differenza (54 kcal/mole) può essere presa come la forza di un secondo legame π. I 9 kcal/mole che indeboliscono questo secondo legame π è riflesso nei numeri del calore di idrogenazione (36.7 - 28.3 = 8.4).
Dato che gli alchini sono termodinamicamente meno stabili degli alcheni, dovremmo aspettarci che le reazioni di addizione dei primi siamo più esotermiche e relativamente più veloci delle equivalenti reazioni degli ultimi. Nel caso dell'idrogenazione catalitica, i soliti catalizzatori per idrogenazione Pt e Pd sono così efficaci nel promuovere l'addizione di idrogeno ad entrambi i doppi ed i tripli legami carbonio-carbonio che l'alchene intermedio formato dall'addizione dell'idrogeno all'alchino non può essere isolato. Un catalizzatore meno efficace, il catalizzatore di Lindlar, preparato disattivando (o avvelenando) il convenzionale catalizzatore al palladio trattandolo con piombo acetato e chinolina, permette di convertire gli alchini in alcheni senza ulteriore riduzione ad un alcano. L'addizione dell'idrogeno è stereoselettivamente syn (cioè il 2-butino dà il cis-2-butene). Una riduzione complementare stereoselettiva in maniera anti può essere ottenuta tramite una soluzione di sodio in ammoniaca liquida, ma ne parleremo poi.
R-C≡C-R + H2/Pd Lindlar → cis R-CH=CH-R
R-C≡C-R + 2 Na/NH3 (liq) → trans R-CH=CH-R + 2 NaNH2
Alcheni ed alchini mostrano una curiosa differenza nel comportamento rispetto alla idrogenazione catalitica. Studi independenti delle velocità di idrogenazione per ogni classe indicano che gli alcheni reagiscono più rapidamente degli alchini. Comunque, l'attenta idrogenazione di un alchino procede esclusivamente verso l'alchene finché il primo non viene consumato, a tale punto l'alchene prodotto è molto rapidamente idrogenato ad un alcano. Questo comportamento è spiegato bene tramite le differenze negli stadi della reazione di idrogenazione. Prima che l'idrogeno possa addizionarsi al legame multiplo l'alchene o l'alchino deve essere adsorbito sulla superficie del catalizzatore. A questo riguardo, la formazione di complessi stabili di platino (e palladio) con gli alcheni sono stati descritti, basti pensare ai già citati altrove sale di Zeise o K[PtCl3(C2H4)]·H2O, ed etilenebis(trifenilfosfino)platino, [(C6H5)3P]2Pt(H2C=CH2). Dato che gli alchini adsorbono più fortemente tali superfici catalitiche rispetto a quanto fanno gli alcheni, essi occupano preferenzialmente i siti reattivi sul catalizzatore. Il successivo transfer di idrogeno all'alchino adsorbito procede lentamente, rispetto al corrispondente transfer di idrogeno alla molecola di alchene adsorbita. Di conseguenza, la riduzione di tripli legami avviene selettivamente ad una velocità moderata, seguita da rapida addizione di idrogeno all'alchene prodotto. Il catalizzatore di Lindlar permette l'adsorbimento e la riduzione di alchini, ma non adsorbe gli alcheni sufficientemente da permettere la loro riduzione.
I seguenti utenti ringraziano quimico per questo messaggio: AAA
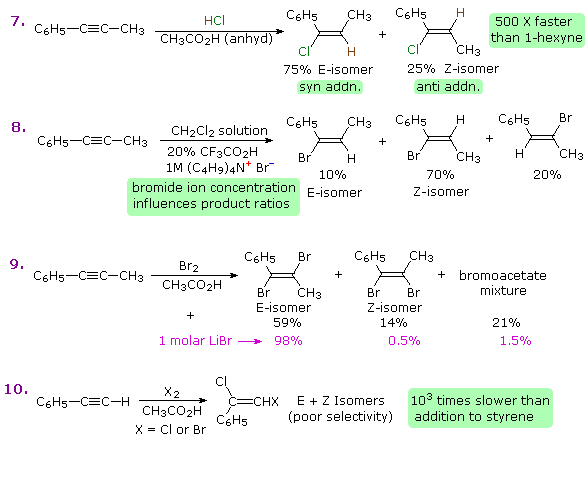
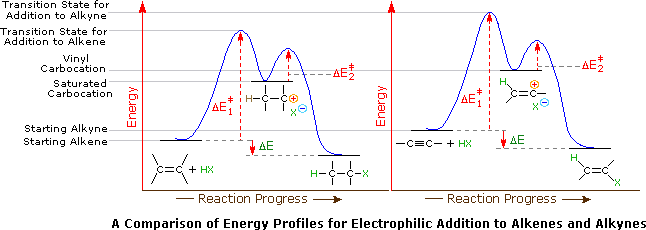
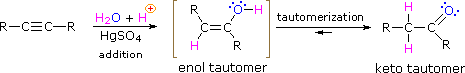
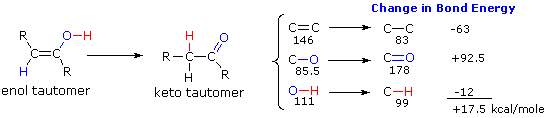
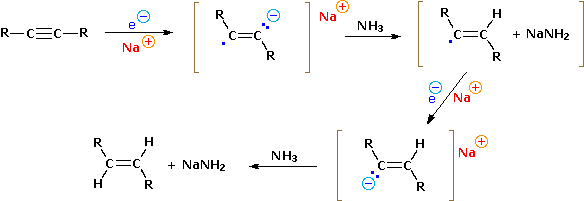
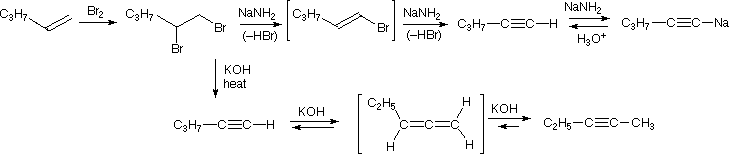 Nel caso degli alchini non terminali, la sodio e la potassio ammide, e le relative basi forti da amine primarie, sono in grado di estrarre protoni da atomi di carbonio adiacenti al triplo legame. I risultanti carbanioni allenici subiscono equilibri rapidi di trasferimento di protone, portando alla base coniugata relativamente stabile dell'alchino terminale. Questa isomerizzazione può essere usata per preparare 1-alchini a catena più lunga, come mostrato nella seguente conversione del 3-eptino ad 1-eptino. I sostituenti R ed R' sull'intermedio allenico vanno dal propile all'idrogeno, non appena avvengono i trasferimenti di protone.
Nel caso degli alchini non terminali, la sodio e la potassio ammide, e le relative basi forti da amine primarie, sono in grado di estrarre protoni da atomi di carbonio adiacenti al triplo legame. I risultanti carbanioni allenici subiscono equilibri rapidi di trasferimento di protone, portando alla base coniugata relativamente stabile dell'alchino terminale. Questa isomerizzazione può essere usata per preparare 1-alchini a catena più lunga, come mostrato nella seguente conversione del 3-eptino ad 1-eptino. I sostituenti R ed R' sull'intermedio allenico vanno dal propile all'idrogeno, non appena avvengono i trasferimenti di protone.